2.2. Replication of Previously Reported Molecular Docking Simulations
The crystal structures of DPP4 and the RBD of SARS-CoV-2 were docked using a rigid molecular docking approach with ZDOCK. However, it was not possible to replicate the binding interactions between DPP4 and the RBD of SARS-CoV-2 predicted by Li et al. [
5]. The binding interactions of the docking pose most similar to the predictions by Li et al. (and indeed having the best interaction energy) are shown in
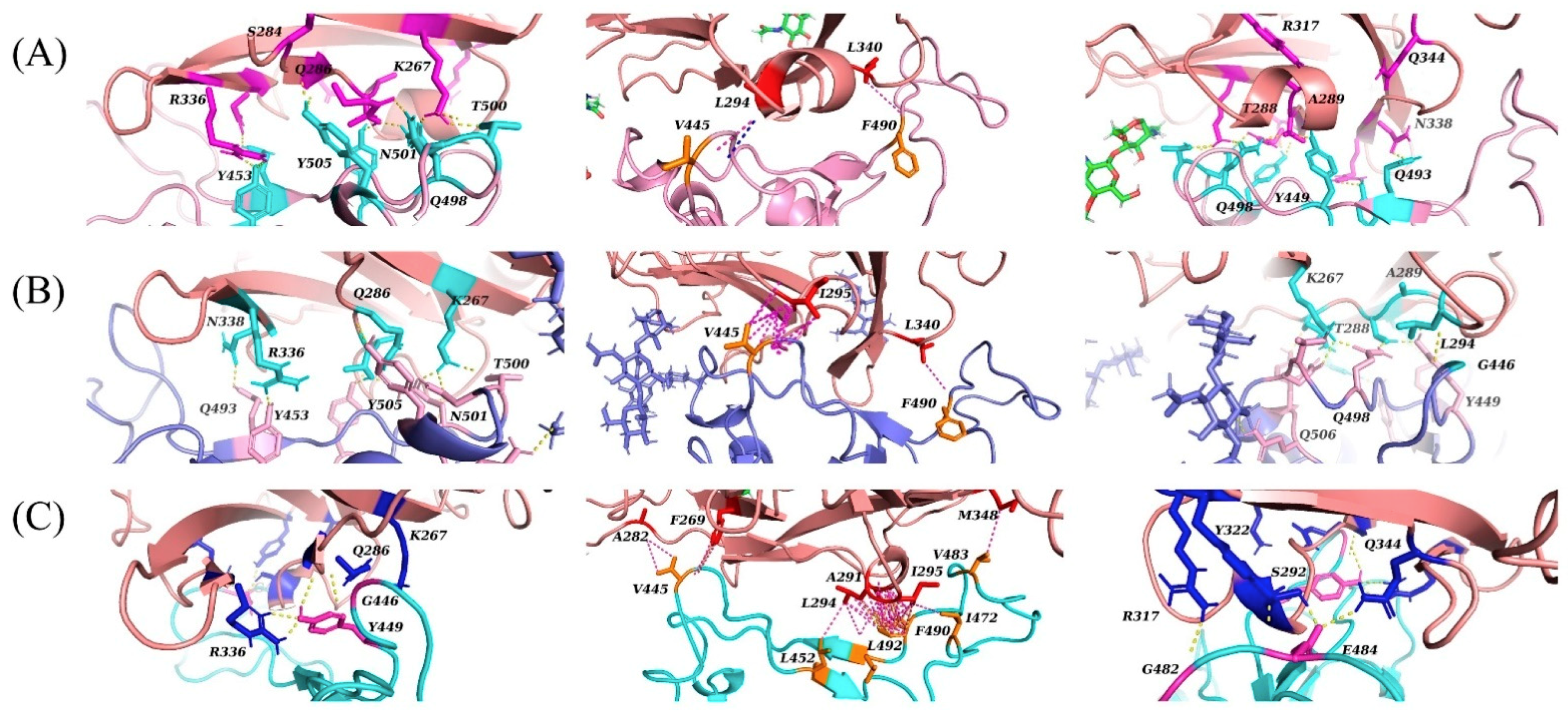
Figure 3. The overall orientation of SARS-CoV-2 appears to have shifted slightly to the right and to the back of the hydrophobic helix of DPP4 compared to the position of MERS-CoV. In comparison to the molecular docking predictions reported by Li et al., this docking pose retains the interactions between the residues R317 and H345 of DPP4 and the residues E484 and N487 of SARS-CoV-2. Interactions that use neighbouring residues to those predicted by Li et al. include the residues K267, R336 and A291 of DPP4, with the residues Y499, E406 and Q493 of SARS-CoV-2. Li et al. predicted that these specific interactions occur between K267 and Q498, R336 and D405, and A289 and Q493 of DPP4 and SARS-CoV-2, respectively. In addition, the following interactions predicted by Li et al. were not reproduced in our simulations: Q344 and Y489, K392 and A475 and T478, Q286 and N501, and T288 and Y505 of DPP4 and SARS-CoV-2, respectively. Overall, the predicted binding interactions bear similarity to the predictions of Li et al. but do not reproduce their findings.
Further attempts were made to replicate these prior predictions using a flexible molecular docking approach with HADDOCK, with three variations in docking restraints (
Supplementary Table S4). For each of these docking variations, the best cluster of binding poses was chosen based on the HADDOCK score and considering only docking poses that were visually similar to the binding position of the RBD of MERS-CoV in its crystal structure complex with DPP4 [
6]. The binding interactions in the best predictions obtained with each one of these three docking approaches are illustrated in
Figure 4.
The first variation in restraints (CS1) aimed to replicate the approach by Li et al. [
5] by only specifying the DPP4 residues that these authors defined as key contact residues, to characterise the influence of conformational flexibility. The resulting docking predictions were not consistent with those reported by Li et al. [
5] or similar to the interactions that we predicted to occur between DPP4 and SARS-CoV-2 based on the MERS-CoV-DPP4 complex (PDB entry 4L72) [
6].
The second variation (CS2) again only specified DPP4 residues but with a different selection (K267, R336, R317, Q344, L294 and I295) [
6,
7]. As would be expected for similar residue restraints, the resulting docking predictions were similar to those predicted in the above first docking variation (CS1).
The third variation (CS3) specified the DPP4 residues described above and also specified the same SARS-CoV-2 residues as those chosen for the remodelled SARS-CoV-2 structure. This approach aimed to partially remove the bias of the crystal structure of the RBD of SARS-CoV-2 towards its conformation when in complex with the ACE2 receptor [
2,
3,
4]. However, the predictions were poor and inconsistent with all other simulations.
Overall, these molecular docking predictions reveal a tendency for the RBD of SARS-CoV-2 in its crystal structure conformation to avoid the hydrophobic α-helix of DPP4. In comparison to the binding mode of MERS-CoV to DPP4, the binding mode of SARS-CoV-2 in these simulations tended to be skewed such that the RBD would lie behind the hydrophobic α-helix. Compared with MERS-CoV, SARS-CoV-2 lacks available hydrophobic residues that would be able to interact with the α-helix of DPP4 and, instead, has a greater number of polar residues in this region. Hydrophobic interactions tended to form on the opposite side and closer to the centre of the RBD, near the β-sheets of SARS-CoV-2. In the first two docking variations, these hydrophobic interactions tended to form between residue V445 of SARS-CoV-2 and the hydrophobic α-helix of DPP4, and between residues F490 and L340 of SARS-CoV-2 and DPP4, respectively. In the third docking variation, however, residue F490, in addition to I472, L491 and L452 of SARS-CoV-2, was predicted to interact with the hydrophobic α-helix of DPP4. Residue V445 was predicted to interact with the DPP4 residues A282 and F269.
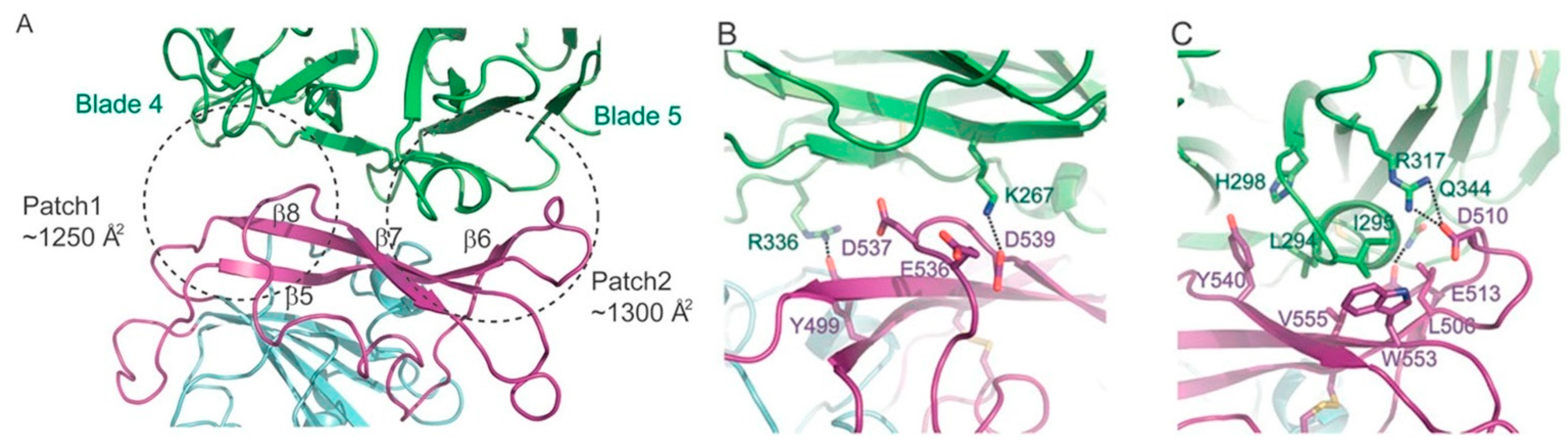
In the MERS-CoV-DPP4 complex, this hydrophobic region of DPP4 is surrounded by the hydrophilic residues R317 and Q344, which interact, respectively, with residues D510 and E513 of MERS-CoV. These residues were not predicted to interact with SARS-CoV-2 in the first two docking variations. In the third variation, however, residue R317 forms a hydrogen bond with G482 (SARS-CoV-2) and Q344 forms a hydrogen bond with E484 (SARS-CoV-2).
In comparison with the binding interactions in the region defined as Patch 1 in the MERS-CoV-DPP4 complex (
Figure 1), only the first two docking variations (CS1 and CS2) resulted in a like-for-like substitution of the DPP4 residue R336. In the crystal structure of the MERS-CoV-DPP4 complex, this residue hydrogen bonds to Y499 of MERS-CoV. In our docking predictions, this residue hydrogen bonds to Y453 of SARS-CoV-2, which is the only sequence-aligned residue of SARS-CoV-2 and MERS-CoV to have an interaction to the correct DPP4 residue. This hydrogen bond was not predicted in the third docking variation (CS3). The salt bridge found in the crystal structure of the MERS-CoV-DPP4 complex with residue K267 of DPP4 was instead replaced with hydrogen bonds. In both CS1 and CS2 variations, this residue tended to interact with T500, N501 and Q498 of SARS-CoV-2. In the CS3 variation, K267 of DPP4 is hydrogen-bonded to G446 of SARS-CoV-2. The DPP4 residue Q286 interacted with residues Y505 and Y499 of SARS-CoV-2 in the first and second, and third variation, respectively.
These findings indicate that the molecular simulations were unable to successfully dock the RBD of SARS-CoV-2 with DPP4 in spite of the conformational flexibility that HADDOCK allowed. This suggested that using a different initial conformation of the RBD of SARS-CoV-2 may be necessary, particularly since the conformation used was extracted from its crystal structure in complex with the ACE2 receptor.
2.3. Molecular Docking of the Remodelled Structure of the RBD of SARS-CoV-2
The best clusters of binding poses for the remodelled structure of the RBD of SARS-CoV-2 were selected based on the requirement that docking poses of the clusters should be visually similar to the RBD of MERS-CoV observed in its crystal structure complex with DPP4 [
6]. The binding interactions between the RBD of SARS-CoV-2 and DPP4 were analysed to identify the cluster with the binding interactions most similar to those at the binding interface of the MERS-CoV-DPP4 complex and/or to eliminate clusters with unlikely interactions. The best cluster was then selected based upon these interactions and the HADDOCK score (
Figure 5). A key difference with respect to the MERS-CoV-DPP4 complex, however, is that the RBD of SARS-CoV-2 was still predicted to be positioned behind the hydrophobic α-helix of DPP4, although to a lesser extent than in the docking predictions using the crystal structure described above.
The best pose selected was ranked favourably by HADDOCK, satisfied interactions with all of the residues in DPP4 specified in the docking restraints and the nature of the interactions with these key residues was reasonable. In comparison to the above-described predicted MERS-CoV-DPP4 complex, the best pose was predicted to have slightly more favourable van der Waals interactions and similar de-solvation energies and buried surface areas (
Table 1). However, the electrostatic interaction was substantially less favourable compared to that predicted in the MERS-CoV-DPP4 complex, which is likely due to the substitution of salt bridges with hydrogen bonds for residues R317 and K267 in DPP4. As a consequence, the binding energy of the interaction of the RBD of SARS-CoV-2 with DPP4 is predicted to be substantially less favourable than that of MERS-CoV. The predicted binding energies using the crystal structure of the RBD of SARS-CoV-2 were worse for all three docking variations in restraints (
Supplementary Table S6).
Table 1 shows that the binding energy of SARS-CoV-2 to ACE2 is predicted to be somewhat more favourable, reflecting of course not only the different interactions of SARS-CoV-2 with each receptor, but also the differences in receptor structures. The de-solvation energy was lower for the SARS-CoV-2-ACE2 complex; however, the van de Waals and electrostatic energies and buried surface area were predicted to be higher than those predicted for the SARS-CoV-2-DPP4 complex.
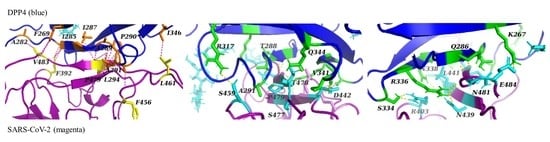
Analysis of this best docking pose revealed that the binding interactions between the remodelled structure of the RDB of SARS-CoV-2 and DPP4 are comparable to the binding interactions in the predicted MERS-CoV-DPP4 complex (
Figure 6) and those observed in the MERS-CoV-DPP4 crystal structure (
Figure 1).
Figure 6 also shows that the docking predictions for the MERS-CoV-DPP4 complex are very similar to the corresponding crystal structure. Consequently, the crystal structure of the MERS-CoV-DPP4 complex was used to compare the docking predictions for the SARS-CoV-DPP4 complex described below.
The SARS-CoV-2-DPP4 complex was predicted to exhibit hydrophobic interactions with a hydrophobic α-helix in DPP4 [
6,
7]. In contrast to MERS-CoV, however, residues L294 and A291 in DPP4 were predicted to interact with residues P479 and F456 in SARS-CoV-2 [
6,
7]. Additionally, P479 interacts with residues I287, A289 and P290 in DPP4. Unlike in the MERS-CoV-DPP4 complex, these hydrophobic interactions do not include I295 in DPP4, and the residues in SARS-CoV-2 do not surround the hydrophobic residues in DPP4 as the residues in MERS-CoV do [
6,
7]. Initially, the residue L461 in SARS-CoV-2 was expected to interact with residues L294 and I195 in DPP4; however, we predicted L461 to instead form a hydrophobic interaction with I346 in DPP4 [
6,
7]. Furthermore, residues A282, F269 and I285 in DPP4 were predicted to have hydrophobic interactions with residues V483 and F392 in SARS-CoV-2 [
6,
7]. The latter were not conserved in all of the docking poses of this cluster.
SARS-CoV-2 was also predicted to exhibit polar interactions with positively charged residues R317 and Q344 in DPP4 that surround its hydrophobic α-helix [
6,
7]. In the MERS-CoV-DPP4 complex, these residues interact with negatively charged acidic amino acid residues; however, in the predicted SARS-CoV-2-DPP4 complex, these residues form hydrogen bonds with S459 and T478 [
6,
7]. As the salt bridge interactions present in the MERS-CoV-DPP4 complex are replaced with only hydrogen bonding, these interactions are arguably much weaker [
6,
7]. However, mutagenesis studies found that these interactions were not significant for the binding of MERS-CoV to DPP4 [
6,
7]. In addition, new hydrogen bonding interactions were predicted between the backbones of residues A291, T288 and V341 in DPP4 and the side chains of residues S477, P479 and D442 in SARS-CoV-2. The interaction between T288 in DPP4 and P479 in SARS-CoV-2 was not conserved in all of the other docking poses of this cluster.
The interactions between SARS-CoV-2 and DPP4 within the second region encompassing polar interactions were found to be reasonable (but weaker) substitutes for the interactions seen in the MERS-CoV-DPP4 complex, in which hydrogen bonding and salt bridges with the amino acid residues R336 and K267 in DPP4 were found to be essential for binding [
6,
7]. In the predicted SARS-CoV-2-DPP4 complex, R366 in DPP4 forms hydrogen bonds with two asparagine residues, N439 and N481. The latter interaction is conserved across half of the four poses, whereas the former interaction is only observed in the best docking pose. This interaction is, however, often observed in other clusters. It is likely that R336 in DPP4 interacts with N481 on the second β-sheet of SARS-CoV-2 because a neighbouring arginine on SARS-CoV-2, R403, repeals R366 in DPP4 to orient this residue closer to a second β-sheet. Residue K267 in DPP4, on the other hand, interacts with residue E484 in SARS-CoV-2. This interaction is deemed to be reasonable as the interaction with an aspartic acid in the MERS-CoV-DPP4 complex is predicted to be replaced with an interaction with a glutamic acid [
6,
7]. Further polar interactions were predicted between residues S334, Q286 and N338 in DPP4 and residues R403, N481 and L441 in SARS-CoV-2. The hydrogen bond between the side chain of N388 and the backbone of L441 is unlikely, however, because leucine has a tendency to interact with other non-polar residues. This interaction was only observed in this docking pose.
Furthermore, these interactions can be compared to those observed in the complex DPP4 forms with adenosine deaminase (ADA). Interestingly, there is a significant cross-over between the DPP4 residues that Weihofen et al. [
16] discuss in the ADA-DPP4 complex and those observed in the predicted interactions in the MERS-CoV-DPP4 and SARS-CoV-2-DPP4 complexes (the latter using the remodelled structure of SARS-CoV-2).
In terms of polar interactions, Weihofen et al. [
16] highlight E139 and D143 in ADA as important residues for binding to DPP4. The residue E139 in ADA hydrogen bonds with S292, A291, P290 and Q344 in DPP4. Hydrogen bonding is also observed between D143 and Q344 of ADA and DPP4, respectively. Interestingly, in the MERS-CoV-DPP4 complex, Q344 in DPP4, one of the polar residues that surrounds the hydrophobic α-helix, hydrogen bonds to E513 in MERS-CoV [
6,
7]. In our remodelled structure, Q344 hydrogen bonds to T478 in SARS-CoV-2. In the MERS-CoV-DPP4 complex, R317 is highlighted as an important residue that forms a salt bridge with the MERS-CoV residue D510 [
6,
7]. In our predictions with the remodelled structure, this interaction is replaced with hydrogen bonding to S459; however, it is absent in the ADA-DPP4 complex. This suggests that the DPP4 residue Q344 may be of greater importance when DPP4 binds to other proteins.
Furthermore, the ADA residue D127 forms a salt bridge to R336 in DPP4. In the MERS-CoV-DPP4 complex, R336 was identified as an important polar residue that hydrogen bonds to the MERS-CoV residue Y499 [
6,
7]. Using the remodelled SARS-CoV-2 structure, R336 was predicted to hydrogen bond with N439 and N481 of SARS-CoV-2. As ADA is able to form a salt bridge with the DPP4 residue R336, this likely improves the binding energy in the ADA-DPP4 complex and again confirms that R336 is an important residue when DPP4 binds to other proteins. It should be noted that a salt bridge was observed between K267 and D539 in DPP4 and MERS-CoV, respectively [
6,
7]. The same interaction was predicted to occur between K267 and E484 in DPP4 and the remodelled SARS-CoV-2, respectively. However, no interaction with K267 in DPP4 is observed in the ADA-DPP4 complex. This suggests that while an interaction with K267 is not necessary for binding to DPP4, strong polar interactions in this area do increase binding to DPP4.
Additional polar interactions in the ADA-DPP4 complex include those between A289 and K80 of DPP4 and ADA [
16]. Hydrogen bonding to this DPP4 residue is not predicted with the remodelled SARS-CoV-2 structure (however, it is in the C1 docking variation); however, the neighbouring DPP4 residue A291 interacts with the remodelled SARS-CoV-2 residue S477. Weihofen et al. [
16] also describe the hydrogen bonding between the DPP4 residues T288 and Q286 and the ADA residue D77. Hydrogen bonding with DPP4 residue Q286 was not observed in the crystal structure of the MERS-CoV-DPP4 complex; however, it was predicted to hydrogen bond to N501 and S559 in MERS-CoV as an additional interaction in our control docking predictions. Hydrogen bonding occurs between T288 and P479, and Q286 and N481, of DPP4 and SARS-CoV-2, respectively. The substitution of non-charged polar residues in the place of the charged ADA residues and hydrogen bonding to proline (the SARS-CoV-2 residue P479) is unlikely to impact binding to DPP4.
Weihofen et al. [
16] reported a number of hydrophobic interactions across the binding interface in the ADA-DPP4 complex. In the ADA-DPP4 complex, the hydrophobic α-helix (DPP4 residues I295, L294, A291 and S292) interacts with Y84 and R81 of ADA. In contrast to ADA, the DPP4 residues I295 and L294 interact with the MERS-CoV residues V555, W553 and L506. Our remodelled structure, discussed above, predicts that L294 and A291 hydrophobically interact with the SARS-CoV-2 residues P479 and F456. The DPP4 residue V341 was also identified as important for binding in the ADA-DPP4 complex [
16,
17]. This residue, along with Q344, was reported to hydrophobically interact with the ADA residues E139 and Q138. These interactions are not present in the MERS-CoV-DPP4 crystal structure; however, hydrophobic interactions were predicted in the docking control for V341 and P515 in DPP4 and MERS-CoV, respectively. No hydrophobic interactions were predicted between V341 and Q344 in DPP4 and the remodelled SARS-CoV-2, respectively; however, these DPP4 residues were involved in hydrogen bonding to SARS-CoV-2.
Both Weihofen et al. [
16] and Abbott et al. [
17] agree that hydrophobic interactions are important for binding through the observation that the ADA-DPP4 complex dissociates at low ionic strength and the point mutations L294R and V341K in DPP4 resulted in a loss of binding. This reflects our conclusions that the inability of SARS-CoV-2 to effectively interact with the hydrophobic α-helix in DPP4 is detrimental to binding.
Additional hydrophobic interactions were reported between the DPP4 residues S292, A291 and P290, and the ADA residue E139 [
16]. These interactions are not observed in the MERS-CoV-DPP4 crystal structure [
6,
7]. However, in the remodelled SARS-CoV-2, the DPP4 residues L294, A291, P290 and A289 hydrophobically interact with P479 in SARS-CoV-2. In the ADA-DPP4 complex, the DPP4 residue I346 was reported to interact with the ADA residues D143 and R142. Interactions with I346 were again not observed in MERS-CoV-DPP4; however, this residue interacts with L461 in SARS-CoV-2. This suggests that, in addition to the residues in the hydrophobic α-helix, the DPP4 residues V341 and I346 should be considered in future predictions of interactions with other proteins.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}