
The Updating of Biological Functions of Methyltransferase SETDB1 and Its Relevance in Lung Cancer and Mesothelioma


Abstract
:1. Introduction
2. Biological Functions of SETDB1
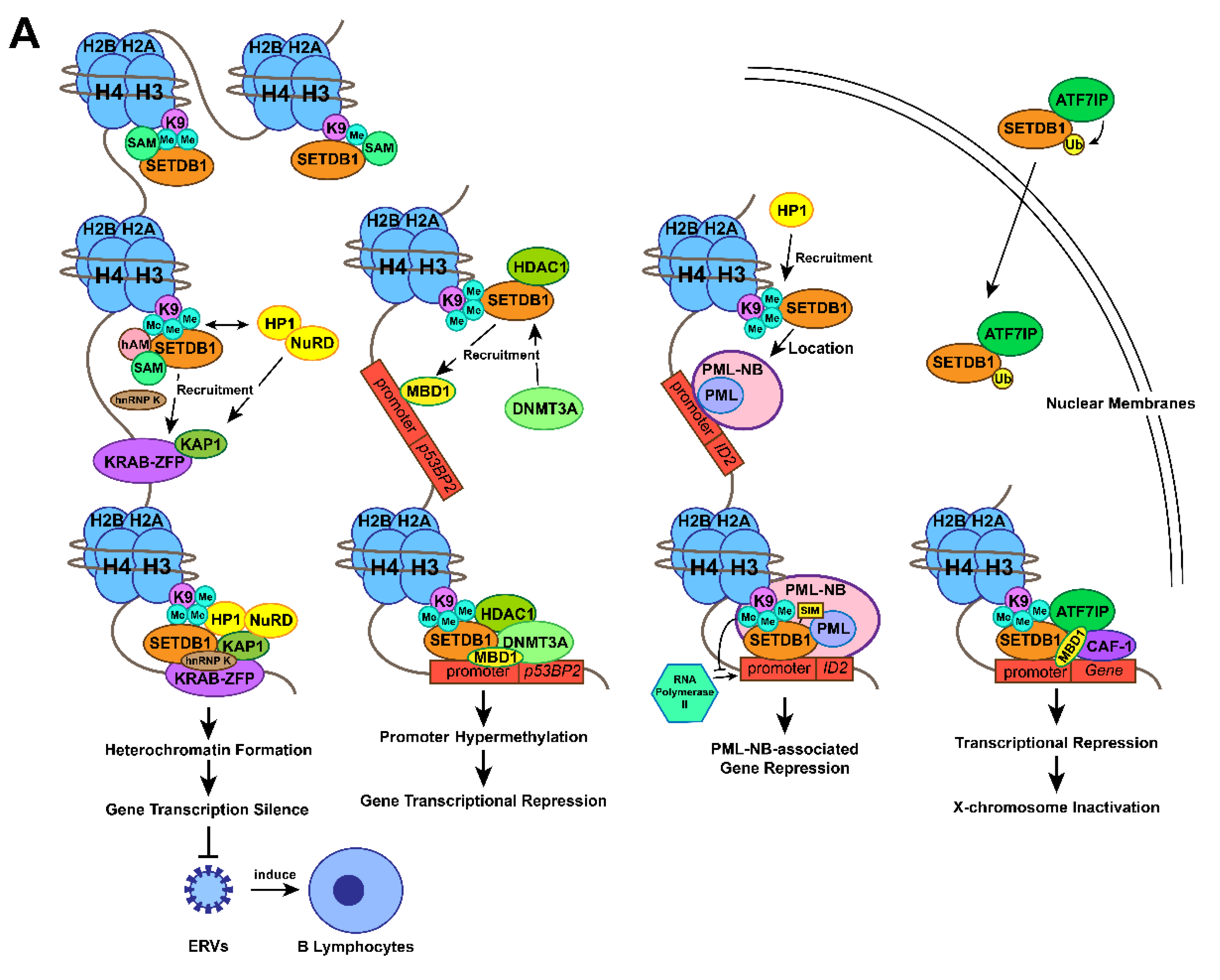
2.1. The Methylation of Histone H3 Lysine 9 by SETDB1
2.2. Gene Transcription Silencing by the SETDB1-KRAB-KAP1 Complex
2.3. Gene Transcription Silencing by Interaction between SETDB1 and DNMT3A
2.4. Gene Transcription Silencing by Interaction between SETDB1 and PML
2.5. X-Chromosome Inactivation by SETDB1-ATF7IP-MBD1 Complex
2.6. Remodelling of Chromatin Associated with SETDB1 Expression
2.7. Early Embryo Development Associated with SETDB1
2.8. Embryonic Stem Cell Development Associated with SETDB1
2.9. Brief Summary of the Functions of SETDB1
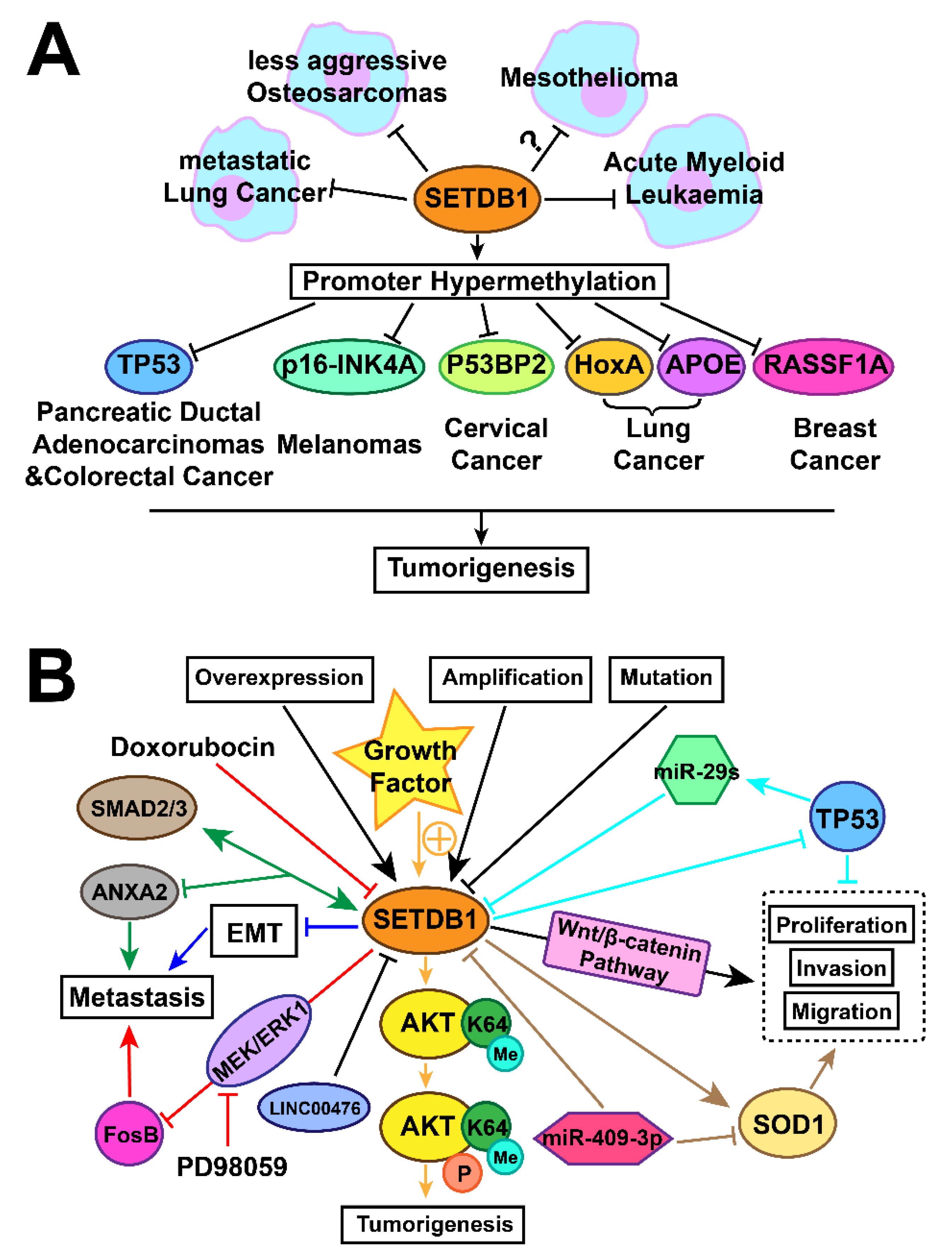
3. Tumourigenesis Associated with Expression of SETDB1
3.1. Lung Cancer
3.1.1. Amplification and Overexpression of SETDB1 in Lung Cancer
3.1.2. SETDB1 Oncoprotein or Tumour-Suppressor Status in Different Stages
3.1.3. Overexpression of SETDB1 Promotes Lung Cancer Growth through Regulating SOD1, LINC00476, p53, and FosB
3.1.4. SETDB1 Inhibits Lung Cancer Metastasis by Regulating SMAD2/3 and EMT
3.2. Malignant Mesothelioma
3.3. Brief Summary of the Regulation Mechanisms of SETDB1 in Cancers with a Focus on Lung Caner
4. Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Rauch, T.; Chen, Z.X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Tapias, P.; Zakharova, V.; Perez-Fernandez, O.M.; Mantilla, W.; RamÍRez-Clavijo, S.; Ait-Si-Ali, S. Expression of the Major and Pro-Oncogenic H3K9 Lysine Methyltransferase SETDB1 in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhuang, S. Histone Methyltransferases as Therapeutic Targets for Kidney Diseases. Front. Pharmacol. 2019, 10, 1393. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Saha, N.; Muntean, A.G. Insight into the multi-faceted role of the SUV family of H3K9 methyltransferases in carcinogenesis and cancer progression. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188498. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; An, W.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Chatton, B.; Tempst, P.; Roeder, R.G.; Zhang, Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 2003, 12, 475–487. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Xiao, J.F.; Chien, W.; Lim, S.L.; Hattori, N.; Goodglick, L.; Chia, D.; Mah, V.; Alavi, M.; et al. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J. Pathol. 2015, 235, 559–570. [Google Scholar] [CrossRef]
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simó-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vázquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813. [Google Scholar] [CrossRef]
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.; Wang, J.; Wang, H.; Gu, X.; Tang, L.; Feng, X. A regulatory circuitry comprising TP53, miR-29 family, and SETDB1 in non-small cell lung cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.H.; Jen, K.Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Emi, M.; Nakano, T.; Gaudino, G. Mesothelioma developing in carriers of inherited genetic mutations. Transl. Lung Cancer Res. 2020, 9, S67–S76. [Google Scholar] [CrossRef]
- Torricelli, F.; Lococo, F.; Di Stefano, T.S.; Lorenzini, E.; Piana, S.; Valli, R.; Rena, O.; Veronesi, G.; Billè, A.; Ciarrocchi, A. Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers 2020, 12, 2454. [Google Scholar] [CrossRef]
- Karanth, A.V.; Maniswami, R.R.; Prashanth, S.; Govindaraj, H.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 319–331. [Google Scholar] [CrossRef]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ye, F.; Li, Y.Y.; Zhan, Y.Z.; Liu, Y.; Yan, H.M.; Fang, Y.; Xie, Y.W.; Zhang, F.J.; Chen, L.H.; et al. Histone methyltransferase SETDB1 promotes colorectal cancer proliferation through the STAT1-CCND1/CDK6 axis. Carcinogenesis 2020, 41, 678–688. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Li, Y.; Quan, C.; Zheng, L.; Huang, K. Lung Cancer Therapy Targeting Histone Methylation: Opportunities and Challenges. Comput. Struct. Biotechnol. J. 2018, 16, 211–223. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Nast, R.; Choepak, T.; Lüder, C.G.K. Epigenetic Control of IFN-γ Host Responses During Infection With Toxoplasma gondii. Front. Immunol. 2020, 11, 581241. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081. [Google Scholar] [CrossRef]
- Rajan, P.K.; Udoh, U.A.; Sanabria, J.D.; Banerjee, M.; Smith, G.; Schade, M.S.; Sanabria, J.; Sodhi, K.; Pierre, S.; Xie, Z.; et al. The Role of Histone Acetylation-/Methylation-Mediated Apoptotic Gene Regulation in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 8894. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Chlamydas, S.; Piperi, C. Histone lysine methyltransferase SETDB1 as a novel target for central nervous system diseases. Prog. Neurobiol. 2021, 200, 101968. [Google Scholar] [CrossRef]
- Kranz, A.; Anastassiadis, K. The role of SETD1A and SETD1B in development and disease. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194578. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- de Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell. Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Minkovsky, A.; Sahakyan, A.; Rankin-Gee, E.; Bonora, G.; Patel, S.; Plath, K. The Mbd1-Atf7ip-Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenet. Chromatin 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Fu, X.; Wang, W.; Chen, X.; Liu, S.; Cao, P.; Zhao, S. LINC00476 Suppresses the Progression of Non-Small Cell Lung Cancer by Inducing the Ubiquitination of SETDB1. Radiat. Res. 2020. [Google Scholar] [CrossRef]
- Na, H.-H.; Kim, K.-C. SETDB1-mediated FosB regulation via ERK2 is associated with an increase in cell invasiveness during anticancer drug treatment of A549 human lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, B.; Hong, L.; Xu, H.; Fishbein, M.C. Epigenetic modifications of histone h4 in lung neuroendocrine tumors. Appl. Immunohistochem. Mol. Morphol. AIMM 2011, 19, 389–394. [Google Scholar] [CrossRef]
- Fischle, W.; Wang, Y.; Allis, C.D. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003, 15, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Smothers, J.F.; Henikoff, S. The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr. Biol. CB 2000, 10, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Ourmouli, N.; Sun, Y.M.; van der Sar, S.; Singh, P.B.; Brown, J.P. Epigenetic regulation of mammalian pericentric heterochromatin in vivo by HP1. Biochem. Biophys. Res. Commun. 2005, 337, 901–907. [Google Scholar] [CrossRef]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J., 3rd. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef]
- Lechner, M.S.; Begg, G.E.; Speicher, D.W.; Rauscher, F.J., 3rd. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: Direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol. 2000, 20, 6449–6465. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef] [Green Version]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell. Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Keown, J.R.; Black, M.M.; Raclot, C.; Demarais, N.; Trono, D.; Turelli, P.; Goldstone, D.C. A Dissection of Oligomerization by the TRIM28 Tripartite Motif and the Interaction with Members of the Krab-ZFP Family. J. Mol. Biol. 2019, 431, 2511–2527. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Geis, F.K.; Goff, S.P. Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses 2020, 12, 884. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Burns, K.H. Mobilizing diversity: Transposable element insertions in genetic variation and disease. Mob. DNA 2010, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mager, D.L.; Stoye, J.P. Mammalian Endogenous Retroviruses. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Shinkai, Y. SETDB1-Mediated Silencing of Retroelements. Viruses 2020, 12, 596. [Google Scholar] [CrossRef]
- Elsässer, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral elements and their hosts: Insertional mutagenesis in the mouse germ line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, B.; Walter, K.; Kreher, S.; Kumar, R.; Hummel, M.; Lenze, D.; Köchert, K.; Bouhlel, M.A.; Richter, J.; Soler, E.; et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med. 2010, 16, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Deniz, Ö.; de la Rica, L.; Cheng, K.C.L.; Spensberger, D.; Branco, M.R. SETDB1 prevents TET2-dependent activation of IAP retroelements in naïve embryonic stem cells. Genome Biol. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Okuda, A.; Yusa, K.; Shinkai, Y. A CRISPR knockout screen identifies SETDB1-target retroelement silencing factors in embryonic stem cells. Genome Res. 2018, 28, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.J.; Dulberg, V.; Moon, K.M.; Foster, L.J.; Chen, C.; Karimi, M.M.; Lorincz, M.C. hnRNP K coordinates transcriptional silencing by SETDB1 in embryonic stem cells. PLoS Genet. 2015, 11, e1004933. [Google Scholar] [CrossRef]
- Collins, P.L.; Kyle, K.E.; Egawa, T.; Shinkai, Y.; Oltz, E.M. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 8367–8372. [Google Scholar] [CrossRef] [Green Version]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Takemoto, K.; Shinkai, Y. A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun. 2018, 9, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Robbez-Masson, L.; Tie, C.H.C.; Rowe, H.M. Cancer cells, on your histone marks, get SETDB1, silence retrotransposons, and go! J. Cell Biol. 2017, 216, 3429–3431. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [Green Version]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021. [Google Scholar] [CrossRef]
- Harjes, U. SETDB1, a new target for immunotherapy. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef]
- Dammann, R.; Schagdarsurengin, U.; Seidel, C.; Strunnikova, M.; Rastetter, M.; Baier, K.; Pfeifer, G.P. The tumor suppressor RASSF1A in human carcinogenesis: An update. Histol. Histopathol. 2005, 20, 645–663. [Google Scholar] [CrossRef]
- Cho, S.; Park, J.S.; Kang, Y.-K. Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): Maintaining PML-NB structure and regulating the expression of its associated genes. J. Biol. Chem. 2011, 286, 41115–41124. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.K. SETDB1 in Early Embryos and Embryonic Stem Cells. Curr. Issues Mol. Biol. 2015, 17, 1–10. [Google Scholar]
- Luciani, J.J.; Depetris, D.; Usson, Y.; Metzler-Guillemain, C.; Mignon-Ravix, C.; Mitchell, M.J.; Megarbane, A.; Sarda, P.; Sirma, H.; Moncla, A.; et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J. Cell Sci. 2006, 119, 2518–2531. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Carter, A.C.; Chang, H.Y. Mechanistic insights in X-chromosome inactivation. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, N.; Bowness, J.S.; Wei, G. Progress toward understanding chromosome silencing by Xist RNA. Genes Dev. 2020, 34, 733–744. [Google Scholar] [CrossRef]
- Keniry, A.; Gearing, L.J.; Jansz, N.; Liu, J.; Holik, A.Z.; Hickey, P.F.; Kinkel, S.A.; Moore, D.L.; Breslin, K.; Chen, K.; et al. Setdb1-mediated H3K9 methylation is enriched on the inactive X and plays a role in its epigenetic silencing. Epigenet. Chromatin 2016, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Tapias, P.; Robin, P.; Pontis, J.; Maestro, L.D.; Ait-Si-Ali, S. The H3K9 Methylation Writer SETDB1 and its Reader MPP8 Cooperate to Silence Satellite DNA Repeats in Mouse Embryonic Stem Cells. Genes 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarraf, S.A.; Stancheva, I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 2004, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Reese, B.E.; Bachman, K.E.; Baylin, S.B.; Rountree, M.R. The methyl-CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Mol. Cell. Biol. 2003, 23, 3226–3236. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, T.; Watanabe, S.; Sakamoto, Y.; Aoto, T.; Fujita, N.; Nakao, M. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J. Biol. Chem. 2005, 280, 13928–13935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsusaka, T.; Shimura, C.; Shinkai, Y. ATF7IP regulates SETDB1 nuclear localization and increases its ubiquitination. EMBO Rep. 2019, 20, e48297. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.A.; Rowe, H.M. A nuclear licence to silence transposons. EMBO Rep. 2019, 20, e49262. [Google Scholar] [CrossRef] [PubMed]
- Tsusaka, T.; Fukuda, K.; Shimura, C.; Kato, M.; Shinkai, Y. The fibronectin type-III (FNIII) domain of ATF7IP contributes to efficient transcriptional silencing mediated by the SETDB1 complex. Epigenet. Chromatin 2020, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Blakeley, P.; Sangrithi, M.N.; Mahadevaiah, S.K.; Encheva, V.; Snijders, A.P.; ElInati, E.; Ojarikre, O.A.; de Rooij, D.G.; Niakan, K.K.; et al. SETDB1 Links the Meiotic DNA Damage Response to Sex Chromosome Silencing in Mice. Dev. Cell 2018, 47, 645–659.e646. [Google Scholar] [CrossRef] [Green Version]
- Navarro, C.; Lyu, J.; Katsori, A.M.; Caridha, R.; Elsässer, S.J. An embryonic stem cell-specific heterochromatin state promotes core histone exchange in the absence of DNA accessibility. Nat. Commun. 2020, 11, 5095. [Google Scholar] [CrossRef]
- Happel, N.; Doenecke, D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene 2009, 431, 1–12. [Google Scholar] [CrossRef]
- Bolderson, E.; Savage, K.I.; Mahen, R.; Pisupati, V.; Graham, M.E.; Richard, D.J.; Robinson, P.J.; Venkitaraman, A.R.; Khanna, K.K. Kruppel-associated Box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1β (HP1-β) mobilization and DNA repair in heterochromatin. J. Biol. Chem. 2012, 287, 28122–28131. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schöfer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Beaujean, N.; Brochard, V.; Audouard, C.; Zink, D.; Debey, P. Genome restructuring in mouse embryos during reprogramming and early development. Dev. Biol. 2006, 292, 317–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antos, F.; Peters, A.H.; Otte, A.P.; Reik, W.; Dean, W. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 2005, 280, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Park, J.S.; Kwon, S.; Kang, Y.K. Dynamics of Setdb1 expression in early mouse development. Gene Expr. Patterns 2012, 12, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; De Falco, F.; Pizzo, M.; Salomoni, P.; Pandolfi, P.P.; Meroni, G. PML interacts with Myc, and Myc target gene expression is altered in PML-null fibroblasts. Oncogene 2005, 24, 2195–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Zhao, H.; Dan, J.; Kim, S.; Hardikar, S.; Hollowell, D.; Lin, K.; Lu, Y.; Takata, Y.; Shen, J.; et al. Maternal Setdb1 Is Required for Meiotic Progression and Preimplantation Development in Mouse. PLoS Genet. 2016, 12, e1005970. [Google Scholar] [CrossRef] [Green Version]
- Eymery, A.; Liu, Z.; Ozonov, E.A.; Stadler, M.B.; Peters, A.H. The methyltransferase Setdb1 is essential for meiosis and mitosis in mouse oocytes and early embryos. Development 2016, 143, 2767–2779. [Google Scholar] [CrossRef] [Green Version]
- Dodge, J.E.; Kang, Y.K.; Beppu, H.; Lei, H.; Li, E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 2004, 24, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, F.; Loureiro, J.; Su, H.; Fang, Q.; Lei, H.; Lewis, T.; Yang, Y.; Labow, M.; Li, E.; Chen, T.; et al. KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells 2010, 28, 201–212. [Google Scholar] [CrossRef]
- Yuan, P.; Han, J.; Guo, G.; Orlov, Y.L.; Huss, M.; Loh, Y.H.; Yaw, L.P.; Robson, P.; Lim, B.; Ng, H.H. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009, 23, 2507–2520. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Liu, H.; Wang, Y.; He, J.; Xu, S.; Chen, Y.; Kuang, J.; Liu, J.; Guo, L.; Li, D.; et al. SETDB1-Mediated Cell Fate Transition between 2C-Like and Pluripotent States. Cell Rep. 2020, 30, 25–36.e26. [Google Scholar] [CrossRef] [Green Version]
- Yeap, L.S.; Hayashi, K.; Surani, M.A. ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage. Epigenet. Chromatin 2009, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Vendetti, F.P.; Rudin, C.M. Epigenetic therapy in non-small-cell lung cancer: Targeting DNA methyltransferases and histone deacetylases. Expert Opin. Biol. Ther. 2013, 13, 1273–1285. [Google Scholar] [CrossRef]
- Bilodeau, S.; Kagey, M.H.; Frampton, G.M.; Rahl, P.B.; Young, R.A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 2009, 23, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Fukuda, A.; Matsumoto, Y.; Hanyu, Y.; Sono, M.; Fukunaga, Y.; Masuda, T.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. SETDB1 Inhibits p53-Mediated Apoptosis and Is Required for Formation of Pancreatic Ductal Adenocarcinomas in Mice. Gastroenterology 2020, 159, 682–696.e613. [Google Scholar] [CrossRef]
- Fei, Q.; Shang, K.; Zhang, J.; Chuai, S.; Kong, D.; Zhou, T.; Fu, S.; Liang, Y.; Li, C.; Chen, Z.; et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015, 6, 8651. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhang, F.; Ding, J.; Liang, Y.; Zhan, Z.; Zhan, Y.; Chen, L.H.; Ding, Y. Histone Methyltransferase SETDB1 Promotes the Progression of Colorectal Cancer by Inhibiting the Expression of TP53. J. Cancer 2017, 8, 3318–3330. [Google Scholar] [CrossRef] [Green Version]
- Kostaki, M.; Manona, A.D.; Stavraka, I.; Korkolopoulou, P.; Levidou, G.; Trigka, E.A.; Christofidou, E.; Champsas, G.; Stratigos, A.J.; Katsambas, A.; et al. High-frequency p16(INK) (4A) promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp. Dermatol. 2014, 23, 332–338. [Google Scholar] [CrossRef]
- Orouji, E.; Federico, A.; Larribère, L.; Novak, D.; Lipka, D.B.; Assenov, Y.; Sachindra, S.; Hüser, L.; Granados, K.; Gebhardt, C.; et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer 2019, 145, 3462–3477. [Google Scholar] [CrossRef]
- Shi, X.; Tasdogan, A.; Huang, F.; Hu, Z.; Morrison, S.J.; DeBerardinis, R.J. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci. Adv. 2017, 3, eaao5268. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.C.; Lu, J.W.; Yang, J.Y.; Lin, I.H.; Ou, D.L.; Lin, Y.H.; Chou, K.H.; Huang, W.F.; Wang, W.P.; Huang, Y.L.; et al. H3K9 histone methyltransferase, KMT1E/SETDB1, cooperates with the SMAD2/3 pathway to suppress lung cancer metastasis. Cancer Res. 2014, 74, 7333–7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafuente-Sanchis, A.; Zúñiga, Á.; Galbis, J.M.; Cremades, A.; Estors, M.; Martínez-Hernández, N.J.; Carretero, J. Prognostic value of ERCC1, RRM1, BRCA1 and SETDB1 in early stage of non-small cell lung cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2016, 18, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 2017, 8, 2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, L.; Robin, P.; Mathieu, J.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Ropa, J.; Saha, N.; Chen, Z.; Serio, J.; Chen, W.; Mellacheruvu, D.; Zhao, L.; Basrur, V.; Nesvizhskii, A.I.; Muntean, A.G. PAF1 complex interactions with SETDB1 mediate promoter H3K9 methylation and transcriptional repression of Hoxa9 and Meis1 in acute myeloid leukemia. Oncotarget 2018, 9, 22123–22136. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.K.; Min, B. SETDB1 Overexpression Sets an Intertumoral Transcriptomic Divergence in Non-small Cell Lung Carcinoma. Front. Genet. 2020, 11, 573515. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung Cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Noh, H.J.; Kim, K.A.; Kim, K.C. p53 down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem Biophys. Res. Commun. 2014, 446, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Al Emran, A.; Marzese, D.M.; Menon, D.R.; Stark, M.S.; Torrano, J.; Hammerlindl, H.; Zhang, G.; Brafford, P.; Salomon, M.P.; Nelson, N.; et al. Distinct histone modifications denote early stress-induced drug tolerance in cancer. Oncotarget 2018, 9, 8206–8222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, A.E.; Halbert, C.L.; Wootton, S.K.; Miller, A.D. Lung cancer in mice induced by the jaagsiekte sheep retrovirus envelope protein is not maintained by rare cancer stem cells, but tumorigenicity does correlate with Wnt pathway activation. Mol. Cancer Res. 2012, 10, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Matsuura, S.; Kurabe, N.; Kahyo, T.; Mori, H.; Kawase, A.; Karayama, M.; Inui, N.; Funai, K.; Shinmura, K.; et al. Clinicopathological and Survival Analysis of Japanese Patients with Resected Non-Small-Cell Lung Cancer Harboring NKX2-1, SETDB1, MET, HER2, SOX2, FGFR1, or PIK3CA Gene Amplification. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Li, B.; Xu, J.; Hu, S.; Zhan, N.; Wang, H.; Gao, C.; Li, J.; Xu, X. SOD1 Promotes Cell Proliferation and Metastasis in Non-small Cell Lung Cancer via an miR-409-3p/SOD1/SETDB1 Epigenetic Regulatory Feedforward Loop. Front. Cell Dev. Biol. 2020, 8, 213. [Google Scholar] [CrossRef]
- Na, H.H.; Noh, H.J.; Cheong, H.M.; Kang, Y.; Kim, K.C. SETDB1 mediated FosB expression increases the cell proliferation rate during anticancer drug therapy. BMB Rep. 2016, 49, 238–243. [Google Scholar] [CrossRef]
- Na, H.H.; Moon, S.; Kim, K.C. Knockout of SETDB1 gene using the CRISPR/cas-9 system increases migration and transforming activities via complex regulations of E-cadherin, β-catenin, STAT3, and Akt. Biochem. Biophys. Res. Commun. 2020, 533, 486–492. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Wong, C.M.; Wei, L.; Law, C.T.; Ho, D.W.; Tsang, F.H.; Au, S.L.; Sze, K.M.; Lee, J.M.; Wong, C.C.; Ng, I.O. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatologists 2016, 63, 474–487. [Google Scholar] [CrossRef] [Green Version]
- Ryu, T.Y.; Kim, K.; Kim, S.K.; Oh, J.H.; Min, J.K.; Jung, C.R.; Son, M.Y.; Kim, D.S.; Cho, H.S. SETDB1 regulates SMAD7 expression for breast cancer metastasis. BMB Rep. 2019, 52, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.V.; Wallen, J.M. Malignant Mesothelioma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Markowitz, P.; Patel, M.; Groisberg, R.; Aisner, J.; Jabbour, S.K.; De, S.; Ganesan, S.; Malhotra, J. Genomic characterization of malignant pleural mesothelioma and associated clinical outcomes. Cancer Treat Res. Commun. 2020, 25, 100232. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Asciak, R.; George, V.; Rahman, N.M. Update on biology and management of mesothelioma. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2021, 30. [Google Scholar] [CrossRef]
- van Gerwen, M.; Alpert, N.; Flores, R.; Taioli, E. An overview of existing mesothelioma registries worldwide, and the need for a US Registry. Am. J. Ind. Med. 2020, 63, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Kanodia, S.; Chao, A.; Miller, A.; Wali, A.; Weissman, D.; Adjei, A.; Baumann, F.; Boffetta, P.; Buck, B.; et al. Consensus Report of the 2015 Weinman International Conference on Mesothelioma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2016, 11, 1246–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2016, 25, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Sugarbaker, D.J.; Richards, W.G.; Bueno, R. Extrapleural pneumonectomy in the treatment of epithelioid malignant pleural mesothelioma: Novel prognostic implications of combined N1 and N2 nodal involvement based on experience in 529 patients. Ann. Surg. 2014, 260, 577–580, discussion 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldini, E.H.; Richards, W.G.; Gill, R.R.; Goodman, B.M.; Winfrey, O.K.; Eisen, H.M.; Mak, R.H.; Chen, A.B.; Kozono, D.E.; Bueno, R.; et al. Updated patterns of failure after multimodality therapy for malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2015, 149, 1374–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, T.K.; Heasley, L.E. Translating mesothelioma molecular genomics and dependencies into precision oncology-based therapies. Semin. Cancer Biol. 2020, 61, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Reguart, N.; Corral, J.; Lianes, P. Malignant pleural mesothelioma: New hope in the horizon with novel therapeutic strategies. Cancer Treat. Rev. 2015, 41, 27–34. [Google Scholar] [CrossRef]
- Pagano, M.; Ceresoli, L.G.; Zucali, P.A.; Pasello, G.; Garassino, M.; Grosso, F.; Tiseo, M.; Soto Parra, H.; Zanelli, F.; Cappuzzo, F.; et al. Mutational Profile of Malignant Pleural Mesothelioma (MPM) in the Phase II RAMES Study. Cancers 2020, 12, 2948. [Google Scholar] [CrossRef]
- Yang, H.; Xu, D.; Schmid, R.A.; Peng, R.W. Biomarker-guided targeted and immunotherapies in malignant pleural mesothelioma. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- Cantini, L.; Hassan, R.; Sterman, D.H.; Aerts, J. Emerging Treatments for Malignant Pleural Mesothelioma: Where Are We Heading? Front. Oncol. 2020, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, L.; Morra, A.; Fonsatti, E.; Cutaia, O.; Fazio, C.; Annesi, D.; Lenoci, M.; Amato, G.; Danielli, R.; Altomonte, M.; et al. Efficacy and safety of an intensified schedule of tremelimumab for chemotherapy-resistant malignant mesothelioma: An open-label, single-arm, phase 2 study. Lancet Respir. Med. 2015, 3, 301–309. [Google Scholar] [CrossRef]
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630. [Google Scholar] [CrossRef]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Hotta, K.; Fujimoto, N.; Kozuki, T.; Aoe, K.; Kiura, K. Nivolumab for the treatment of unresectable pleural mesothelioma. Expert Opin. Biol. Ther. 2019, 20, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Scherpereel, A.; Wallyn, F.; Albelda, S.M.; Munck, C. Novel therapies for malignant pleural mesothelioma. Lancet Oncol. 2018, 19, e161–e172. [Google Scholar] [CrossRef]
- Chia, P.L.; Scott, A.M.; John, T. Epidermal growth factor receptor (EGFR)-targeted therapies in mesothelioma. Expert Opin. Drug Deliv. 2019, 16, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Adusumilli, P.S.; Alexander, H.R., Jr.; Baas, P.; Bardelli, F.; Bononi, A.; Bueno, R.; Felley-Bosco, E.; Galateau-Salle, F.; Jablons, D.; et al. Mesothelioma: Scientific clues for prevention, diagnosis, and therapy. CA Cancer J. Clin. 2019, 69, 402–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukuyan, A.M.; Sementino, E.; Kadariya, Y.; Menges, C.W.; Cheung, M.; Tan, Y.; Cai, K.Q.; Slifker, M.J.; Peri, S.; Klein-Szanto, A.J.; et al. Inactivation of Bap1 Cooperates with Losses of Nf2 and Cdkn2a to Drive the Development of Pleural Malignant Mesothelioma in Conditional Mouse Models. Cancer Res. 2019, 79, 4113–4123. [Google Scholar] [CrossRef] [PubMed]
- Badhai, J.; Pandey, G.K.; Song, J.-Y.; Krijgsman, O.; Bhaskaran, R.; Chandrasekaran, G.; Kwon, M.-C.; Bombardelli, L.; Monkhorst, K.; Grasso, C.; et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J. Exp. Med. 2020, 217, e20191257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Sato, A.; Tsujimura, T.; Otsuki, T.; Fukuoka, K.; Hasegawa, S.; Nakano, T.; Hashimoto-Tamaoki, T. Biallelic germline and somatic mutations in malignant mesothelioma: Multiple mutations in transcription regulators including mSWI/SNF genes. Int. J. Cancer 2015, 136, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, A.B.; Mitsunaga, S.I.; Cheng, J.Q.; Klein, W.M.; Jhanwar, S.C.; Seizinger, B.; Kley, N.; Klein-Szanto, A.J.; Testa, J.R. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc. Natl. Acad. Sci. USA 1995, 92, 10854–10858. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.Q.; Jhanwar, S.C.; Klein, W.M.; Bell, D.W.; Lee, W.C.; Altomare, D.A.; Nobori, T.; Olopade, O.I.; Buckler, A.J.; Testa, J.R. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994, 54, 5547–5551. [Google Scholar] [PubMed]
- Jean, D.; Daubriac, J.; Le Pimpec-Barthes, F.; Galateau-Salle, F.; Jaurand, M.C. Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch. Pathol. Lab. Med. 2012, 136, 277–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkqvist, A.M.; Tammilehto, L.; Anttila, S.; Mattson, K.; Knuutila, S. Recurrent DNA copy number changes in 1q, 4q, 6q, 9p, 13q, 14q and 22q detected by comparative genomic hybridization in malignant mesothelioma. Br. J. Cancer 1997, 75, 523–527. [Google Scholar] [CrossRef] [Green Version]
- de Reyniès, A.; Jaurand, M.C.; Renier, A.; Couchy, G.; Hysi, I.; Elarouci, N.; Galateau-Sallé, F.; Copin, M.C.; Hofman, P.; Cazes, A.; et al. Molecular classification of malignant pleural mesothelioma: Identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1323–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Histone Methyltransferase | Modification Type | Links to Cancer | References |
|---|---|---|---|
| MLL2 | H3K4me1 | Loss and deleterious mutations in NSCLC; Haematopoietic malignancies | [23,24] |
| SET1B | H3K4me3 | Regulation of transcription to maintain stem cell identity | [3,24,25,26] |
| ET1A | H3K4me1/me2/me3 | [26] | |
| ASH1 | H3K4me1/me2/me3 | [26] | |
| G9a | H3K9me1/me2 | Overexpression of G9a promotes metastasis of lung cancer cells | [23,26] |
| SUV39H2 | H3K9me2/me3 | Promotion of the proliferation and metastasis in intestinal cancer cells; Inhibition of gastric cancer and lung cancer | [8,26] |
| SETDB1 | H3K9me2/me3 | Promotion of liver cancer; CNS diseases | [26,27,28] |
| SETDB2 | H3K9me1/me2/me3 | Inhibition of gastric cancer and haematologic malignancies | [8,26] |
| CLL8 | H3K9me1/me2/me3 | Obesity and fatty liver | [26,27] |
| EZH2 | H3K27me1/me2/me3 | Overexpression in lung cancer; Malignant tumours of the haematopoietic system and hepatocellular carcinoma | [3,23,27,29] |
| SMYD2 | H3K36me1/me2/me3 | Proliferation of lung cancer through ALK activation; Contribution to NSCLC cell growth | [23] |
| SETD2 | H3K36me1/me2/me3 | Inhibition of lung cancer; Harmful mutations in primary NSCLC | [23] |
| WHSC1LI | H3K36me1/me2/me3 | Overexpression in lung cancer | [23] |
| SET3 | H3K36me1/me2/me3 | [26] | |
| NSD1/2/3 | H3K36me1/me2/me3 | [26] | |
| DOT1L | H3K79me1/me2/me3 | Promotion of NSCLC cell growth | [3,23] |
| SUV4-20H1/2 | H4K20me3 | Decreased H4K20me3 in tumour progression | [23] |
| NSD1 | H4K20me1/me2/me3 | [26] | |
| PRMTs | Arginine on H3 and H4 | Overexpression in TKI resistant NSCLC promotes NSCLC growth | [23] |
| Mutation Modalities | Nucleotide Alterations | Protein Alterations | Mutation Location | Effects | References |
|---|---|---|---|---|---|
| Artificial single-amino acid substitution (Base substitution) | H1224K | C-terminal (SET domain) | Impair histone H3 methylase activity; accelerate melanoma | [9,108] | |
| C1226A | C-terminal (SET domain) | Impair H3 methylase activity; accelerate melanoma | [9,108] | ||
| C1279Y | C-terminal (SET domain) | Impair H3-methylase activity | [9] | ||
| Spontaneous mutations in MPM patients (Base substitution) | 2606G>A Missense | G869E | C-terminal (Bifurcated SET) | (Damaging) | [17] |
| 2840C>G Missense | S947C | C-terminal (Bifurcated SET) | Unknown | [17] | |
| 2732G>T Missense | C911F | C-terminal (Bifurcated SET) | Unknown | [17] | |
| 747T>A Nonsense | Y249X | N-terminal | Loss of function; Potential role in MPM development | [17] | |
| Spontaneous mutations in MPM patients (Deletion mutations) | 677_693del17 Frameshift (duplicate) | P226RfsX4 | N-terminal | Loss of function (duplicate mutation) | [17] |
| P226RfsX4 | N-terminal | [17] | |||
| 3747_3749del In-frame deletion | F1250del | C-terminal (Post-SET) | Unknown | [17] | |
| 2020delA In-frame deletion | K674SFSX73 | C-terminal (Between Pre-SET and MBD domian) | Loss of function | [163] | |
| 395_399del5 Frameshift | V132FS | N-terminal | Loss of function; premature stop codon in MPM development | ACCMESO1 mesothelioma cell line (Cancer Cell Line Encyclopedia (CCLE) database) |
| Study Design and Subjects | Conclusion | Reference |
|---|---|---|
| Primary tumours of lung cancer patients at different grades (n = 192) and adjacent normal tissues (n = 16) | SETDB1 overexpression in lung cancer (especially in early ones) | [115] |
| Primary NSCLC at Stage I (n = 64) and adjacent normal tissues | Poor prognostic marker of high SETDB1 mRNA in early NSCLC | [116] |
| Eight microarrays from GEO and Expression Atlas Databases; Primary NSCLC (n = 60) and their paired adjacent normal tissues (n = 60) | SETDB1 overexpression in NSCLC (especially in advanced ones) | [11] |
| Lung cancer tissues (n = 387) and normal bronchial epithelium cells (n = 106) | SETDB1 overexpression in NSCLC (especially in advanced ones) | [11] |
| Primary ADC (n = 164) and SCC (n = 99) tissues | High-level SETDB1 gene amplification in ADC tissues (especially in advanced ones); poor survival marker of SETDB1 gene amplification in ADC; low-level SETDB1 gene amplification in SCC tissues | [128] |
| Primary ADC (n = 20), SCC (n = 20), SCLC (n = 19) tissues | Gene amplification and high protein level of SETDB1 in NSCLC and SCLC | [12] |
| TCGA ADC dataset | Gene amplification and high protein level of SETDB1 in NSCLC | [11] |
| NSCLC (n = 1140) and controls (n = 952) | High expression of SETDB1 in NSCLC | [2] |
| 12 microarray datasets of NSCLC patients, including current smoker (n = 297), former smoker (n = 547) and non-smoker (n = 220) | Higher SETDB1 mRNA expression in patients with smoking history | [2] |
| NSCLC tissues in cBioPortal and Oncomine database (n = 1926) | High SETDB1 mRNA expression in NSCLC (a poor prognostic marker) | [13] |
| Primary NSCLC tissues (n = 9) and paired adjacent normal tissues (n = 9) | SETDB1 overexpression in NSCLC | [13] |
| Primary NSCLC tissues (n = 156) | High SETDB1 expression in NSCLC | [13] |
| 34 microarray datasets of ADC and SCC patients | Association between SETDB1 expression and TP53 mutations in NSCLC | [2] |
| Primary NSCLC tissues (n = 30) in Stage III and IV and paired adjacent normal tissues (n = 30) | SETDB1 mRNA upregulation in primary NSCLC tissues; negative correlation between SETDB1 and TP53 mRNA levels | [14] |
| Primary MPM tissues samples with no prior systemic therapy (n = 74) from The Cancer Genome Atlas (TCGA); Japanese International Cancer Genome Consortium (ICGC) MPM cohort (n = 80) | Inactivation of SETDB1 in MPM; a novel genomic subtype with TP53 and SETDB1 mutations and extensive loss of heterozygosity | [16] |
| Transcriptomes (n = 211), whole exomes (n = 99), whole-genome (n = 1) and targeted exomes (n = 103) from human primary MPM tumour tissues (n = 217) | Recurrent and significant SETDB1 mutations in MPM | [15] |
| MPM patients enrolled in the Ramucirumab Mesothelioma clinical trial (RAMES) (n = 110) | SETDB1 mutation in MPM | [15] |
| 78 MPM tissues from MPM patients (n = 69) | Frequent SETDB1 mutations in MPM | [17] |
| Genetic variation of Japanese with mesothelioma based on the exome sequencing (n = 1208) and on genotyping data of common variations (n = 3248) from the Human Genetic Variation Database (HGVD) | Germline variants and rare missense variants of SETDB1 in mesothelioma | [18] |
| A multicentric retrospective case-control cohort of surgically resected MPMs (n = 69) | SETDB1 mutations in epithelioid and biphasic MPM | [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Sun, B.; Xu, L.; Chen, L.; Ou, W. The Updating of Biological Functions of Methyltransferase SETDB1 and Its Relevance in Lung Cancer and Mesothelioma. Int. J. Mol. Sci. 2021, 22, 7416. https://doi.org/10.3390/ijms22147416
Yuan L, Sun B, Xu L, Chen L, Ou W. The Updating of Biological Functions of Methyltransferase SETDB1 and Its Relevance in Lung Cancer and Mesothelioma. International Journal of Molecular Sciences. 2021; 22(14):7416. https://doi.org/10.3390/ijms22147416
Chicago/Turabian StyleYuan, Li, Boshu Sun, Liangliang Xu, Limin Chen, and Wenbin Ou. 2021. "The Updating of Biological Functions of Methyltransferase SETDB1 and Its Relevance in Lung Cancer and Mesothelioma" International Journal of Molecular Sciences 22, no. 14: 7416. https://doi.org/10.3390/ijms22147416
APA StyleYuan, L., Sun, B., Xu, L., Chen, L., & Ou, W. (2021). The Updating of Biological Functions of Methyltransferase SETDB1 and Its Relevance in Lung Cancer and Mesothelioma. International Journal of Molecular Sciences, 22(14), 7416. https://doi.org/10.3390/ijms22147416