
Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective






Abstract
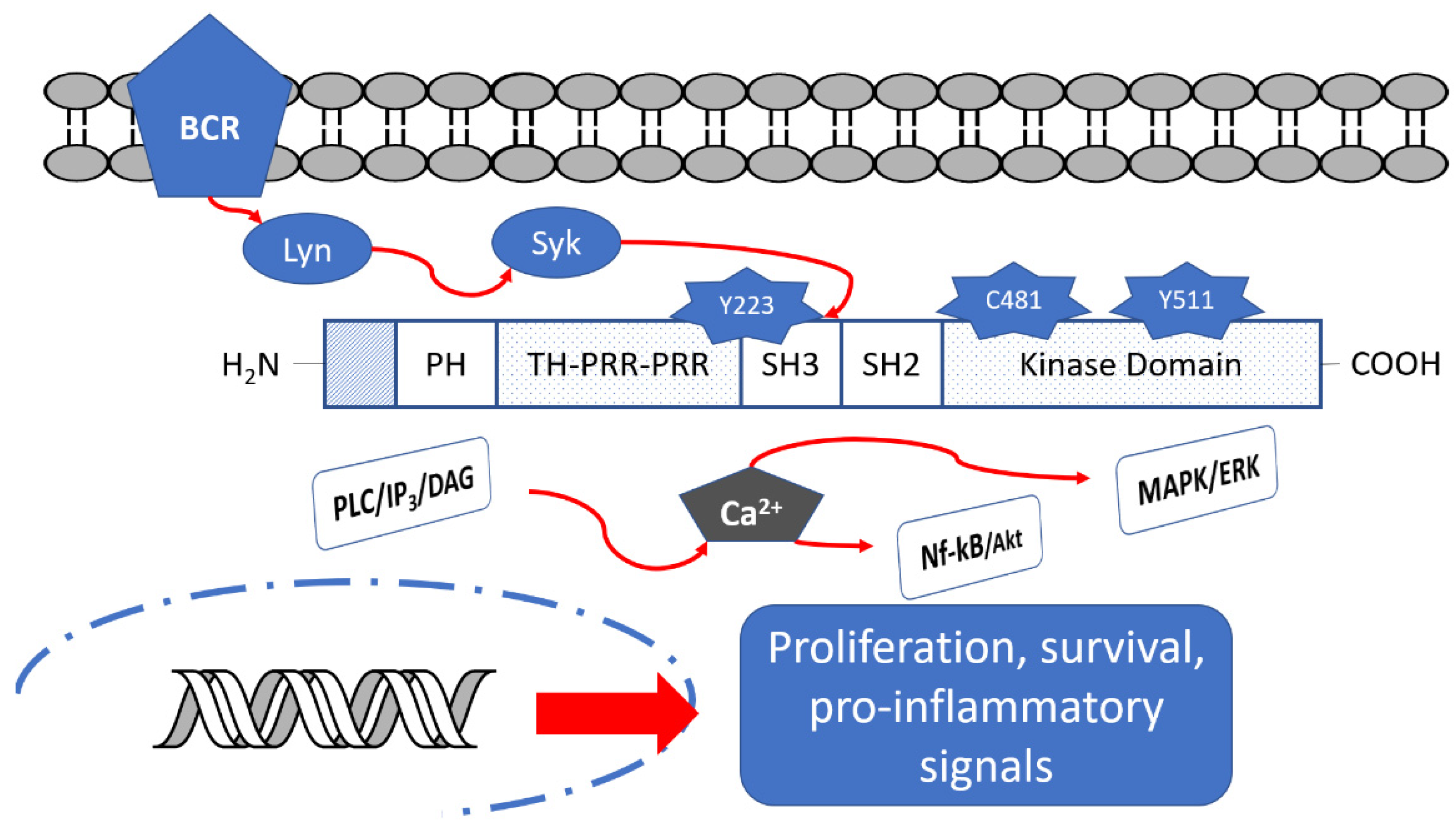
:1. Introduction
2. Btk Structure
3. Btk Inhibitors
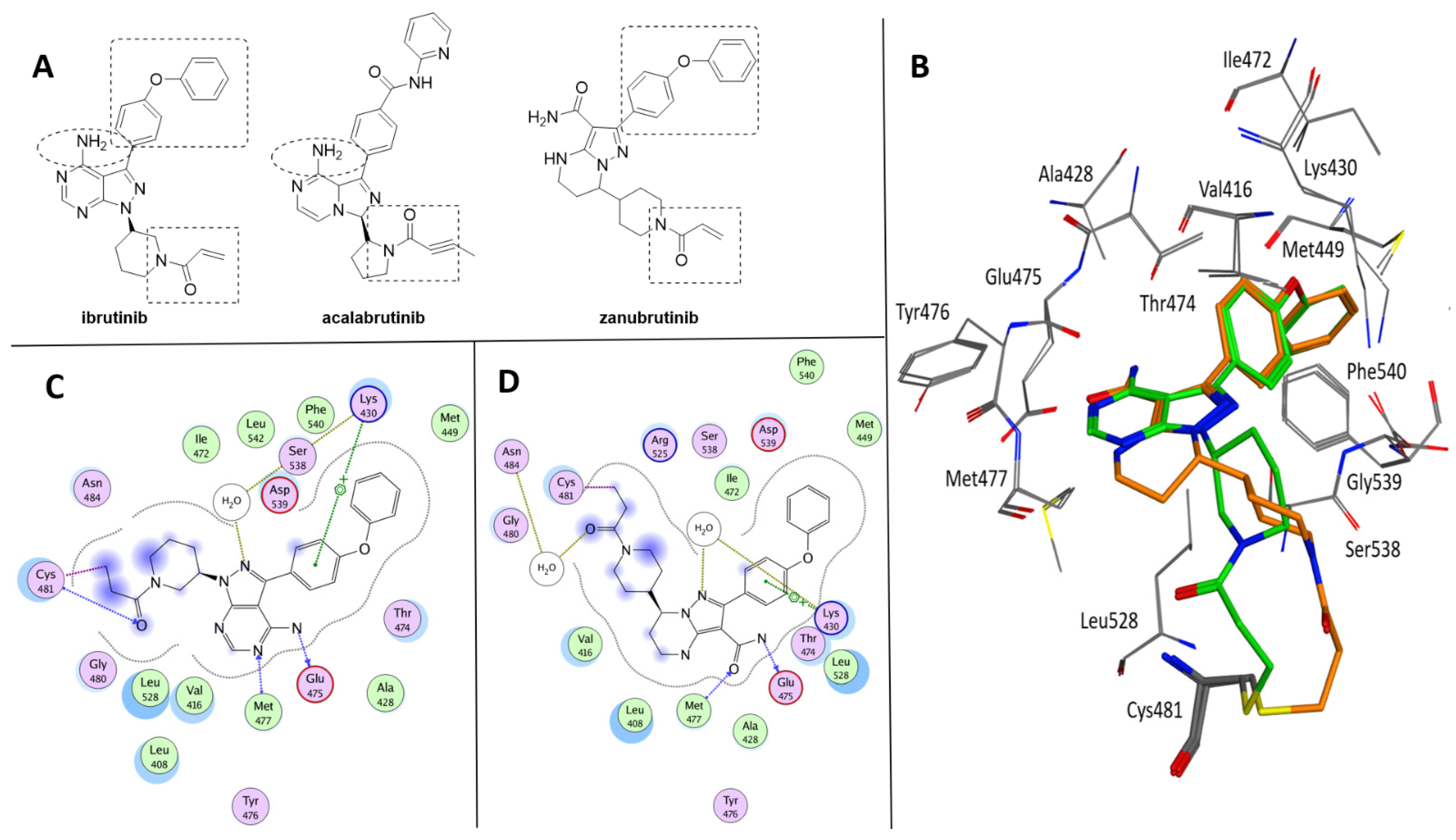
3.1. Approved Btk Inhibitors
Drug Delivery of Ibrutinib
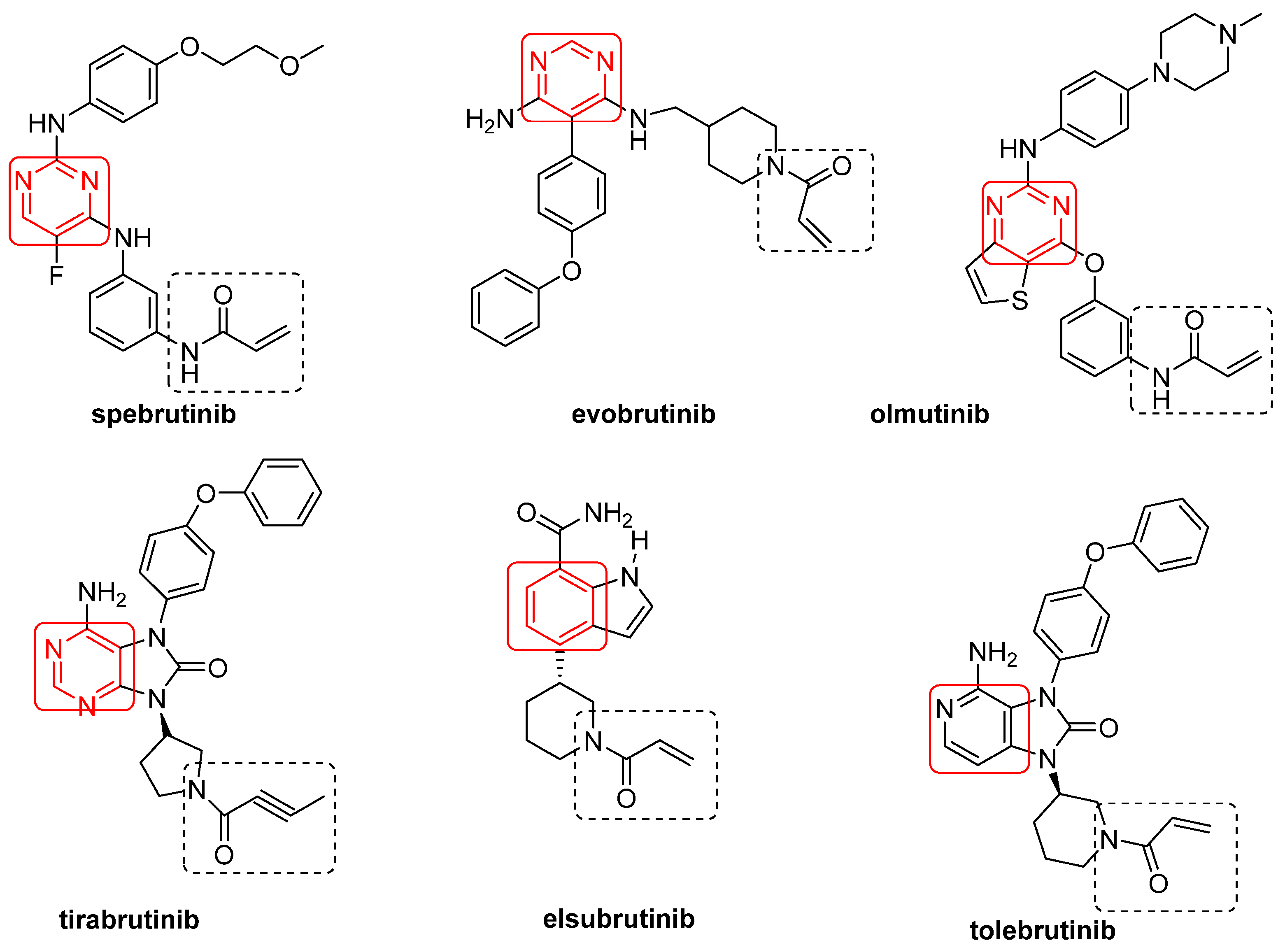
3.2. BtkIs under Clinical Investigation
3.2.1. Irreversible BtkIs
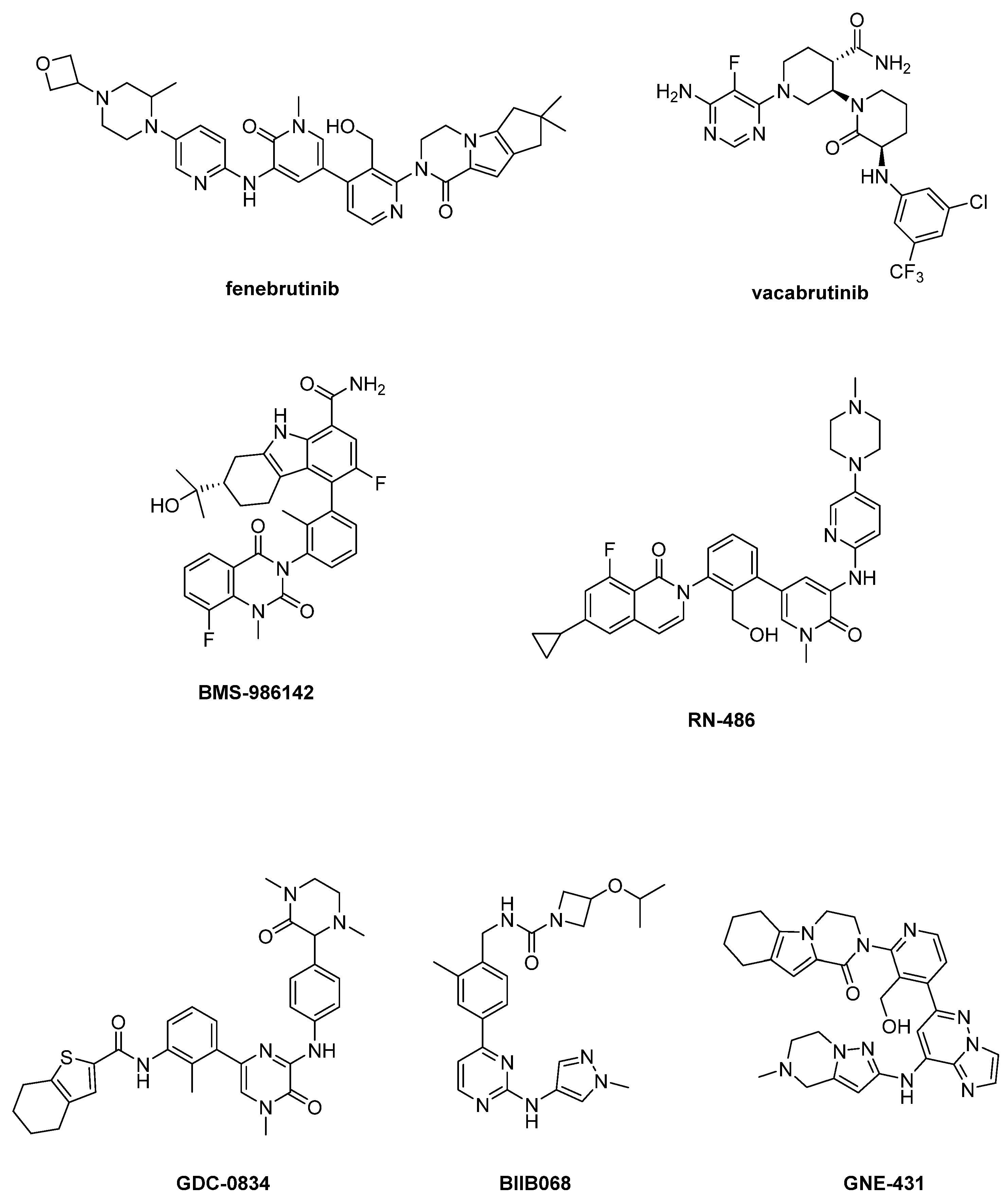
3.2.2. Reversible BtkIs
3.2.3. Emerging Reversible Covalent BtkIs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AERhB | Aminoethyl rhodamine |
| BCR | B cell receptor |
| Btk | Bruton’s kinase |
| BtkIs | Btk inhibitors |
| cGVHD | Chronic graft versus host disease |
| CLL | Chronic lymphocytic leukemia |
| COVID-19 | Coronavirus Disease 19 |
| DAC | 2,3-dialdehyde cellulose |
| EGFR | Epidermal growth factor receptor |
| MCL | Mantle cell lymphoma |
| MS | Multiple sclerosis |
| MSRR | Relapsing-Remitting Multiple Sclerosis |
| MZL | Marginal zone lymphoma |
| NP | Nanoparticles |
| NSCL | Non-Small Cell Lung Cancer |
| PH | Pleckstrin homology domain |
| PROTAC | Proteolysis targeting chimera |
| PRR | Proline-rich regions |
| RA | Rheumatoid arthritis |
| RMS | Relapsing Multiple Sclerosis |
| SH2/SH3 | Src homology 2/3 domain |
| SLE | Systemic Lupus Erythematosus |
| SLL | Small lymphocytic lymphoma |
| Syk | Spleen tyrosine kinase |
| TH | Tec homology domain |
| TK | Tyrosine kinase |
| WM | Waldenström’s macroglobulinemia |
| XLA | X-linked agammaglobulinemia |
References
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarström, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Buggy, J.J.; Elias, L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int. Rev. Immunol. 2012, 31, 119–132. [Google Scholar] [CrossRef]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Hendriks, R.W.; de Bruijn, M.F.; Maas, A.; Dingjan, G.M.; Karis, A.; Grosveld, F. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. Embo. J. 1996, 15, 4862–4872. [Google Scholar] [CrossRef] [PubMed]
- Kil, L.P.; de Bruijn, M.J.; van Nimwegen, M.; Corneth, O.B.J.; Piet van Hamburg, J.; Dingjan, G.M.; Thaiss, F.; Rimmelzwaan, G.F.; Elewaut, D.; Delsing, D.; et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 2012, 119, 3744–3756. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012-2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Dingjan, G.M.; Middendorp, S.; Dahlenborg, K.; Maas, A.; Grosveld, F.; Hendriks, R.W. Bruton’s tyrosine kinase regulates the activation of gene rearrangements at the lambda light chain locus in precursor B cells in the mouse. J. Exp. Med. 2001, 193, 1169–1178. [Google Scholar] [CrossRef] [Green Version]
- Ysebaerta, L.; Michallet, A.S. Bruton’s tyrosine kinase inhibitors: Lessons learned from bench-to-bedside (first) studies. Curr. Opin. Oncol. 2014, 26, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Carnero Contentti, E.; Correale, J. Bruton’s tyrosine kinase inhibitors: A promising emerging treatment option for multiple sclerosis. Expert Opin. Emerg. Drugs 2020, 25, 377–381. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2020, in press. [Google Scholar] [CrossRef]
- Wang, B.; Deng, Y.; Chen, Y.; Yu, K.; Wang, A.; Liang, Q.; Wang, W.; Chen, C.; Wu, H.; Hu, C.; et al. Structure-activity relationship investigation for benzonaphthyridinone derivatives as novel potent Bruton’s tyrosine kinase (BTK) irreversible inhibitors. Eur. J. Med. Chem. 2017, 137, 545–557. [Google Scholar] [CrossRef]
- Yoon, Y. Small chemicals with inhibitory effects on PtdIns(3,4,5)P 3 bindingof Btk PH domain. Bioorg. Med. Chem. Lett. 2014, 24, 2334–2339. [Google Scholar] [CrossRef]
- Jiang, Z.; Liang, Z.; Shen, B.; Hu, G. Computational analysis of the binding specificities of PH domains. Biol. Med. Res. Int. 2015, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Simar, P.S.; Floris, D.; Rudi, W.H. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Zuo, Y.; Pan, Z. Small-Molecule Inhibitors of Bruton’s Tyrosine Kinase. In Cancer II; Topics in Medicinal Chemistry; Waring, M.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 28, pp. 75–104. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s tyrosine kinase inhibitors prevent therapeutic escape in breast cancer cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.M.; Foley, S.E.; Goedken, E.R.; Michalak, M.; Murdock, S.; Wilson, N.S. Merits and Pitfalls in the Characterization of Covalent Inhibitors of Bruton’s Tyrosine Kinase. SLAS Discov. 2018, 23, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.R.; Kohli, P.B.; Katewa, A.; Gogol, E.; Belmont, L.D.; Choy, R.; Penuel, E.; Burton, L.; Eigenbrot, C.; Yu, C.; et al. Battling btk mutants with non covalent inhibitors that overcome Cys481 and Thyr474 mutation. ACS Chem. Biol. 2016, 11, 2897–2907. [Google Scholar] [CrossRef]
- Liu, L.; Shi, B.; Wan, X.; Xiang, H. Strategies to overcome resistance mutations of Bruton’s tyrosine kinase inhibitor ibrutinib. Future Med. Chem. 2018, 10, 343–356. [Google Scholar] [CrossRef]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients with Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Duan, W.; Cu, X.; Xin, M. Bruton’s tyrosine kinase (BTK) inhibitors in treating cancer: A patent review. Expert Opin. Ther. Pat. 2019, 29, 217–241. [Google Scholar] [CrossRef]
- Norman, P. Investigational Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin. Investig. Drugs 2016, 25, 891–899. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Lithgow, H.; Dittus, L.; Bassi, Z.I.; Hughes, S.E.; Muelbaier, M.; Dai, H.; Smith, I.E.D.; Kerr, W.J.; Burley, G.A.; et al. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem. Biol. 2019, 14, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancher, K.M.; Pappacena, J.J. Drug interactions with Bruton’s tyrosine kinase inhibitors: Clinical implications and management. Cancer Chemother. Pharm. 2020, 86, 507–515. [Google Scholar] [CrossRef]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), A novel, potent, And selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Ghosh, S.; Sudbeck, E.A.; Zheng, Y.; Downs, S.; Hupke, M.; Uckun, F.M. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton’s tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2, 5-dibromophenyl)propenamide]. J. Biol. Chem. 1999, 274, 9587–9599. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Schutt, S.D.; Fu, J.; Nguyen, H.; Bastian, D.; Heinrichs, J.; Wu, Y.; Liu, C.; Mcdonald, D.G.; Pidala, J.; Yu, X.Z. Inhibition of BTK and ITK with ibrutinib is effective in the prevention of chronic graft-versus-host disease in mice. PLoS ONE 2015, 10, e0137641. [Google Scholar] [CrossRef] [Green Version]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.; Kath, J.C.; Niessen, S.; et al. A road map toevaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibaud, V.; Beylot-Barry, M.; Protin, C.; Vigarios, E.; Recher, C.; Ysebaert, L. Dermatological Toxicities of Bruton’s Tyrosine Kinase Inhibitors. Am. J. Clin. Dermatol. 2020, 21, 799–812. [Google Scholar] [CrossRef]
- Tillman, B.F.; Pauff, J.M.; Satyanarayana, G.; Talbott, M.; Warner, J.L. Systematic review of infectious events with the Bruton tyrosine kinase inhibitor ibrutinib in the treatment of hematologic malignancies. Eur. J. Haematol. 2018, 100, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, M.P.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tsakmaklis, N.; Yang, G.; Chen, J.G.; Liu, X.; Demos, M.; Kofides, A.; Patterson, C.J.; Meid, K.; Gustine, J.; et al. Acquired mutations associated with ibrutinib resistance in Waldenstrom macroglobulinemia. Blood 2017, 129, 2519–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha-Bang, C.; Niemann, C.U. Targeting Bruton’s Tyrosine Kinase Across B-Cell Malignancies. Drugs 2018, 78, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Harrington, B.K.; Gardner, H.L.; Izumi, R.; Hamdy, A.; Rothbaum, W.; Coombes, K.R.; Covey, T.; Kaptein, A.; Gulrajani, M.; Van Lith, B.; et al. Preclinical Evaluation of the Novel BTK Inhibitor Acalabrutinib in Canine Models of B-Cell Non-Hodgkin Lymphoma. PLoS ONE 2016, 11, e159607. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Kim, E.S.; Eaby, B.; Garey, J.; West, D.P.; Lacouture, M.E. Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: An evolving paradigm in clinical management. Oncologist 2007, 12, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, A.; de Bruin, G.; Emmelot-van Hoek, M.; van de Kar, B.; de Jong, A.; Gulrajani, M.; Demont, D.; Covey, T.; Mittag, D.; Barf, T. Potency and selectivity of BTK inhibitors in clinical development for B-Cell malignancies. Blood 2018, 132, 1871. [Google Scholar] [CrossRef]
- Flinsenberg, T.W.H.; Tromedjo, C.C.; Hu, N.; Liu, Y.; Guo, Y.; Thia, K.Y.T.; Noori, T.; Song, X.; Yeang, H.X.A.; Tantalo, D.G.; et al. Differential effects of BTK inhibitors ibrutinib and zanubrutinib on NK cell effector function in patients with mantle cell lymphoma. Haematologica 2020, 105, 76–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.; Sadaram, M.; Hebb, J.; Sagiv-Barfi, I.; Ambulkar, S.; Rajapaksa, A.; Chang, S.; Chester, C.; Waller, E.; Lai Wang, L.; et al. Three BTK-specific inhibitors, in contrast to ibrutinib, do not antagonize rituximab-dependent NK-cell mediated cytotoxicity. Blood 2014, 124, 3118. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Sun, X.; Mcauley, E.M.; Hsieh, M.M.; Pittaluga, S.; Raffeld, M.; Liu, D.; Keyvanfar, K.; Chapman, C.M.; Chen, J. Modeling tumorehost interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia 2013, 27, 2311–2321. [Google Scholar] [CrossRef] [Green Version]
- Torke, S.; Webera, M.S. Inhibition of Bruton’s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Expert Opin Investig. Drugs 2020, 29, 1143–1150. [Google Scholar] [CrossRef]
- Chong, E.A.; Roeker, L.E.; Shadman, M.; Davids, M.S.; Schuster, S.J.; Mato, A.R. BTK Inhibitors in Cancer Patients with COVID-19: The Winner Will be the One Who Controls That Chaos (Napoleon Bonaparte). Clin. Cancer Res. 2020, 26, 3514–3516. [Google Scholar] [CrossRef] [PubMed]
- Acalabrutinib Study with Best Supportive Care in Participants Hospitalized with COVID-19. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04497948 (accessed on 16 June 2021).
- Svenson, S. What nanomedicine in the clinic right now really forms nanoparticles? WIREs Nanomed. Nanobiotechnol. 2014, 6, 125–135. [Google Scholar] [CrossRef]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Rink, J.S.; Yang, S.; Cen, O.; Taxter, T.; McMahon, K.M.; Misener, S.; Behdad, A.; Longnecker, R.; Gordon, L.I.; Thaxton, C.S. Rational targeting of cellular cholesterol in diffuse large B-Cell Lymphoma (DLBCL) enabled by functional lipoprotein nanoparticles: A therapeutic strategy dependent on cell of origin. Mol. Pharm. 2017, 14, 4042–4051. [Google Scholar] [CrossRef]
- Sanchez-Coronilla, A.; Martin, E.I.; Fernandez-de-Cordova, F.J.; Prado-Gotor, R.; Hidalgo, J. Theoretical study on the interactions between ibrutinib and gold nanoparticles for being used as drug delivery in the chronic lymphocytic leukemia. J. Mol. Liq. 2020, 316, 113878. [Google Scholar] [CrossRef]
- Peng, X.; Liu, P.; Pang, B.; Yao, Y.; Wang, J.; Zhang, K. Facile fabrication of pH-responsive nanoparticles from cellulose derivatives via Schiff base formation for controlled release. Carbohydr. Polym. 2019, 216, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Li, C.; Zhang, H.; Hu, L.; Yan, X.; Zheng, H.; Liu, M.; Liu, M.; Liu, M.; Liu, X. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019, 92, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Rangaraj, N.; Pailla, S.R.; Chowta, P.; Sampathi, S. Fabrication of ibrutinib nanosuspension by quality by design approach: Intended for enhanced oral bioavailability and diminished fast fed variability. AAPS Pharm. Sci. Tech. 2019, 20, 1–18. [Google Scholar] [CrossRef]
- Alshetaili, A.S.; Ansari, M.J.; Anwer, K.; Ganaie, M.A.; Iqbal, M.; Alshahrani, S.M.; Alalaiwe, A.S.; Alsulays, B.B.; Alshehri, S.; Sultan, A.S. Enhanced oral bioavailability of ibrutinib encapsulated poly (lactic-co- glycolic acid) nanoparticles: Pharmacokinetic evaluation in rats. Curr. Pharm. Anal. 2019, 15, 661–668. [Google Scholar] [CrossRef]
- Gao, C.; Chen, F.; Wang, H. Patent analysis of Bruton’s tyrosine kinase inhibitor ibrutinib. Guoji Yaoxue Yanjiu Zazhi 2020, 47, 16–24. [Google Scholar] [CrossRef]
- Chen, W.; Loury, D.J.; Mody, T.D. Preparation of N-{3-[2-(phenylamino)pyrimidin-4-ylamino]phenyl} Amides as Inhibitors of Bruton’s Tyrosine Kinase. WO 2013173518 A1, 21 November 2013. [Google Scholar]
- Hodous, B.L.; Liu-Bujalski, L.; Jones, R.; Bankston, D.; Johnson, T.L.; Mochalkin, I.; Nguyen, N.; Qiu, H.; Goutopoulos, A.; Brugger, N. Compositions and Methods for the Production of Pyrimidine and Pyridine Compounds with Btk Inhibitory Activity. WO 2012170976 A2, 13 December 2012. [Google Scholar]
- Wang, Y.; Li, H. Preparation of Fused Pyrimidine Compound as Protein Kinase Inhibitor. WO 2017092523 A1, 8 June 2017. [Google Scholar]
- Zhao, X.; Xin, M.; Wang, Y.; Huang, W.; Jin, Q.; Tang, F.; Wu, G.; Zhao, Y.; Xiang, H. Discovery of thieno[3,2-c]pyridin-4-amines as novel Bruton’s tyrosine kinase (BTK) inhibitors. Bioorg. Med. Chem. 2015, 23, 6059–6068. [Google Scholar] [CrossRef]
- Bonafoux, D.; Davis, H.M.; Frank, K.E.; Friedman, M.M.; Herold, J.M.; Hoemann, M.Z.; Huntley, R.; Osuma, A.; Sheppard, G.; Somal, G.K.; et al. Preparation of Heterocycle Carboxamides as Bruton’s Tyrosine Kinase (Btk) Inhibitors. WO 2014210255 A1, 31 December 2014. [Google Scholar]
- Goess, C.; Harris, C.M.; Murdock, S.; McCarthy, R.W.; Sampson, E.; Twomey, R.; Mathieu, S.; Mario, R.; Perham, M.; Goedken, E.R.; et al. ABBV-105, A selective and irreversible inhibitor of Bruton’s tyrosine kinase, Is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. 2019, 29, 510–522. [Google Scholar] [CrossRef]
- Cai, X.; Qian, C.; He, Q.; Huang, Y.; Ma, Z.; Qin, S.; Ye, C.; Zhong, X. Preparation of Dihydroimidazopyridine Derivatives for Treatment of Bruton’s Tyrosine Kinase Related Diseases. CN 105753863 A, 13 July 2016. [Google Scholar]
- Dhillon, S. Tirabrutinib: First approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef]
- Kim, E.S. Olmutinib: First global approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef]
- Liu, B.; Shun, Y.T.; Zhou, Y.; Zhang, Y.; Zheng, C. Preparation of 4-amino-3-(alkylethynyl)-1H-pyrazolo[3,4-d]pyrimidine Derivatives Useful as BTK Inhibitors. CN 107344940 A, 14 November 2017. [Google Scholar]
- Mato, A.R.; Schuster, S.J.; Foss, F.M.; Isufi, I.; Ding, W.; Brander, D.M.; Sitlinger, A.; Tun, H.W.; Moustafa, M.A.; Kennard, K.; et al. A Phase Ia/Ib Study Exploring the Synthetic Lethality of the Orally Administered Novel BTK Inhibitor, Dtrmwxhs-12 (DTRM-12), in Combination with Everolimus and Pomalidomide in Patients with Relapsed/Refractory CLL, DLBCL or Other B-Cell Lymphomas. Blood 2019, 134, 810. [Google Scholar] [CrossRef]
- He, W. Preparation of Fluorinated Pyrazolopyrimidine Bruton’s Tyrosine Kinase Inhibitors, Their Drug Combinations and Their Pharmaceutical Compositions and Their Use for Treatment of Cancer and Autoimmune Diseases. US 20160324878 A1, 10 November 2016. [Google Scholar]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein. kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A. Bruton’s tyrosine kinase (BTK) inhibitors in clinical trials. Curr. Hematol. Malig. Rep. 2014, 9, 44–49. [Google Scholar] [CrossRef]
- Spaargaren, M.; de Rooij, M.F.; Kater, A.P.; Eldering, E. BTK inhibitors in chronic lymphocytic leukemia: A glimpse to the future. Oncogene 2015, 34, 2426–2436. [Google Scholar] [CrossRef]
- Crawford, J.J.; Ortwine, D.F.; Wei, B.; Young, W.B. Heteroarylpyridone and Azapyridone Compounds as Inhibitors of BTK Activity and Their Preparation. WO 2013067274 A1, 10 May 2013. [Google Scholar]
- Thompson, P.A.; Burger, J.A. Bruton’s tyrosine kinase inhibitors: First and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL). Expert Opin. Investig. Drugs 2018, 27, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.T.; Conlon, P.; Chan, T.R.; Jenkins, T.J.; Cai, X.; Humora, M.; Shi, X.; Miller, R.A.; Thompson, A. Preparation of Pyrimidinyl Bipiperidine Compounds as Bruton’s Tyrosine Kinase Inhibitors Useful in Treatment of Disease. WO 2013185084 A1, 12 December 2013. [Google Scholar]
- Batt, D.G.; Bertrand, M.B.; Delucca, G.; Galella, M.A.; Ko, S.S.; Langevine, C.M.; Liu, Q.; Shi, Q.; Srivastava, A.S.; Anurag, S.; et al. Substituted Carbazolecarboxamide Derivatives as Btk Inhibitors and Their Preparation and Use for the Treatment of Disease. USA 20140378475 A1, 25 December 2014. [Google Scholar]
- Watterson, S.H.; De Lucca, G.V.; Shi, Q.; Langevine, C.M.; Liu, Q.; Batt, D.G.; Beaudoin, B.M.; Gong, H.; Dai, J.; Yip, S.; et al. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J. Med. Chem. 2016, 59, 9173–9200. [Google Scholar] [CrossRef]
- Berthel, S.; Firooznia, F.; Fishlock, D.; Hong, J.B.; Lou, Y.; Lucas, M.; Owens, T.D.; Sarma, K.; Sweeney, Z.K.; Taygerly, J.P.G. Preparation of 5-phenyl-1H-pyridin-2-one, 6-phenyl-2H-pyridazin-3-one, and 5-phenyl-1H-pyrazin-2-one Derivatives as Inhibitors of Bruton’s Tyrosine Kinase. USA 20100222325 A1, 2 September 2010. [Google Scholar]
- Lou, Y.; Han, X.; Kuglstatter, A.; Kondru, R.K.; Sweeney, Z.K.; Soth, M.; McIntosh, J.; Litman, R.; Suh, J.; Kocer, B.; et al. Structure-based drug design of RN486, a potent and selective Bruton’s tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. J. Med. Chem. 2015, 58, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Halladay, J.S.; Shin, Y.; Wong, S.; Coraggio, M.; La, H.; Baumgardner, M.; Le, H.; Gopaul, S.; Boggs, J.; et al. Significant species difference in amide hydrolysis of GDC-0834, a novel potent and selective Bruton’s tyrosine kinase inhibitor. Drug Metab. Dispos. 2011, 39, 1840–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, W.B.; Barbosa, J.; Blomgren, P.; Bremer, M.C.; Crawford, J.J.; Dambach, D.; Gallion, S.; Hymowitz, S.G.; Kropf, J.E.; Lee, S.H.; et al. Potent and selective Bruton’s tyrosine kinase inhibitors: Discovery of GDC-0834. Bioorg. Med. Chem. Lett. 2015, 25, 1333–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.T.; Ma, B.; Chan, T.R.; Sun, L.; Zhang, L.; Kumaravel, G.; Lyssikatos, J.P.; Koch, K.; Miao, H. Preparation of Pyrazolylaminopyrimidinylbenzylthiazolecarboxamide Derivatives and Analogs for Use as Bruton’s Tyrosine Kinase Inhibitors. WO 2015089337 A1, 18 June 2015. [Google Scholar]
- Ma, B.; Bohnert, T.; Otipoby, K.L.; Tien, E.; Arefayene, M.; Bai, J.; Bajrami, B.; Bame, E.; Chan, T.R.; Humora, M.; et al. Discovery of BIIB068: A Selective, Potent, Reversible Bruton’s Tyrosine Kinase Inhibitor as an Orally Efficacious Agent for Autoimmune Diseases. J. Med. Chem. 2020, 63, 12526–12541. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.J.; Ortwine, D.F.; Young, W.B. Bicyclic Piperazine Compounds as BTK Inhibitors and Their Preparation. WO 2013067260 A1, 10 May 2013. [Google Scholar]
- Smith, F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Gourlay, S.G. SAT0232 a phase 1 clinical trial of PRN1008, an oral, reversible, covalent BTK inhibitor demonstrates clinical safety and therapeutic levels of BTK occupancy without sustained systemic exposure. Ann. Rheum. Dis. 2015, 74, 742. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Owens, T.; Verner, E. Preparation of Pyrazolopyrimidine Compounds as Tyrosine Kinase Inhibitors for Treating Cancer, Autoimmune Disorders, and Inflammation. WO 2014039899 A1, 13 March 2014. [Google Scholar]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nature Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
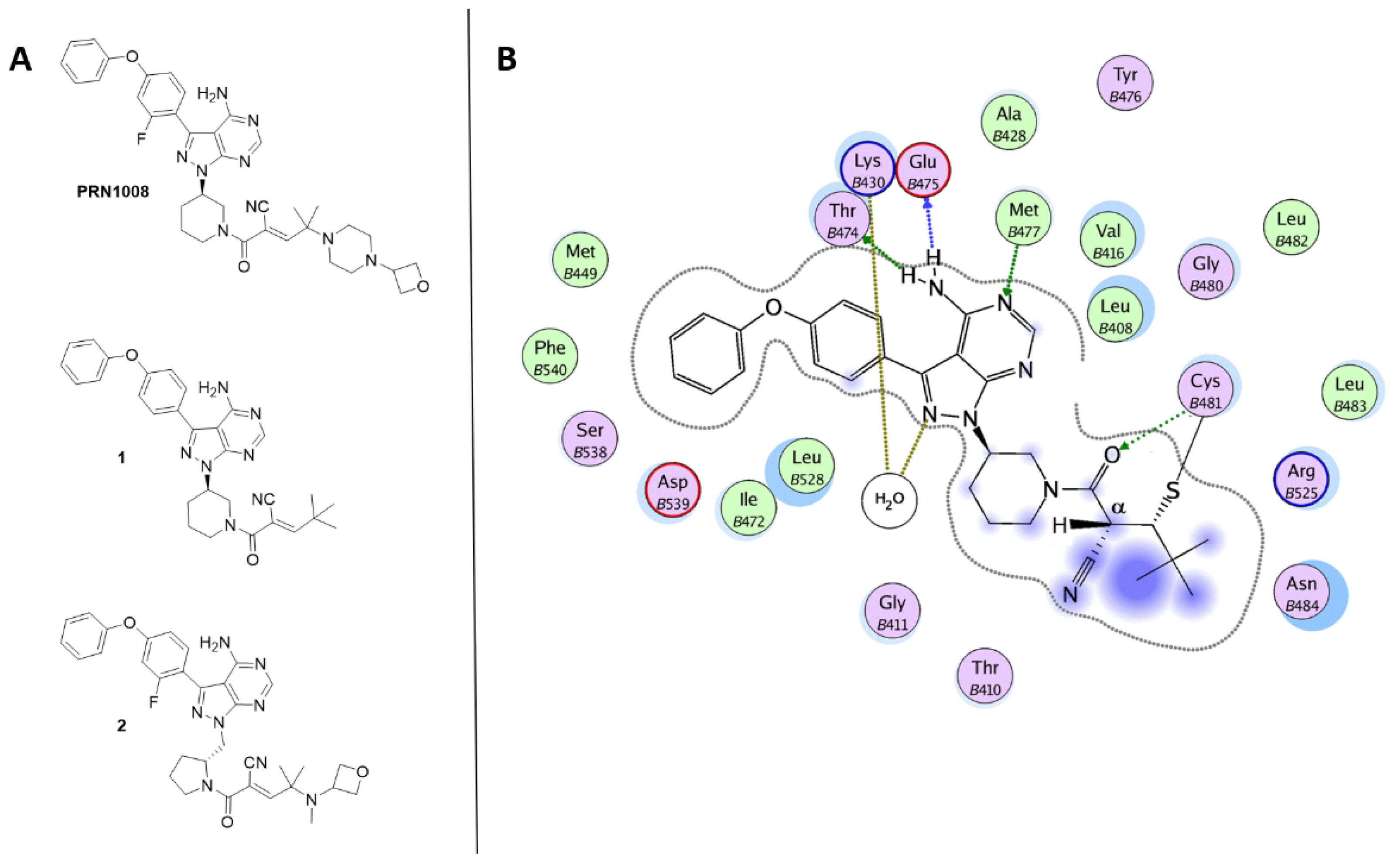{kind=link}
| Drug | Dosage | Diseases | Number of Clinical Trials | ||
|---|---|---|---|---|---|
| Total | GVHD | COVID-19 | |||
| Ibrutinib | 420 mg/day (CLL/SLL, WM) 560 mg/day (MCL, MZL) | CLL/SLL WM MZL MCL cGVHD | 358 | 9 (1 completed, 5 in recruitment, 2 actives not recruiting, 1 enrolling by invitation) | 2 (NCT04375397 in recruitment; NCT04665115 not recruiting) |
| Acalabrutinib | 100 mg b.i.d. | CLL/SLL MCL | 99 | 2 (NCT04198922, NCT04716075, in recruitment) | 3 (NCT04497948 terminated, NCT04380688 completed, NCT04346199 active not recruitment) |
| Zanubrutinib | 160 mg b.i.d. | MCL | 49 | none | 1 (NCT04382586 in recruitment) |
| Compound | No. of Clinical Trials | Treated Diseases |
|---|---|---|
| Spebrutinib | 8 (7 completed, 1 active, not in recruitment) | B Cell Non-Hodgkin’s Lymphoma, CLL, WM, RA |
| Evobrutinib | 15 (4 in recruitment, the others completed or terminated) | MS, MSRR, RMS, RA, SLE |
| Olmutinib | 8 (completed or terminated) | Different adenocarcinoma, NSCL, RA |
| Tirabrutinib | 9 | CLL, SLL, Non-Hodgkin’s Lymphoma, WM, RA, Sjogren’s Syndrome |
| Elsubrutinib | 2, in recruitment | SLE |
| Tolebrutinib | 7 (2 completed) | RMS, primary and secondary progressive MS |
| Fenebrutinib | 8 (3 completed) | Urticaria, SLE, RMS, progressive MS, Lymphocytic Leukemia, lymphoma |
| Vacabrutinib | 1 (terminated) | SLL, CLL |
| PRN1008 (Rilzabrutinib) | 5 (1 completed) | RA, Immune Thrombocytopenia, Immune Thrombocytopenic Purpura, Pemphigus vulgaris |
| BMS-986142 | 7 (all terminated or completed) | RA, Sjögren’s Syndrome |
| CT-1530 | 1 | Lymphoma, different autoimmune diseases |
| TG-1701 | 1 (recruiting) | Non-Hodgkin’s Lymphoma, CLL |
| AC0058 | 2 (1 completed) | SLE |
| SHR1459 | 7 (2 completed) | Lymphoma, different autoimmune diseases |
| RN-486 | Preclinical investigation | |
| BIIB068 | 1 (completed) | SLE |
| DTRMWXHS-12 | 4 (2 completed, 1 in recruitment, 1 active not recruiting) | CLL and different leukemia and lymphoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. Int. J. Mol. Sci. 2021, 22, 7641. https://doi.org/10.3390/ijms22147641
Brullo C, Villa C, Tasso B, Russo E, Spallarossa A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. International Journal of Molecular Sciences. 2021; 22(14):7641. https://doi.org/10.3390/ijms22147641
Chicago/Turabian StyleBrullo, Chiara, Carla Villa, Bruno Tasso, Eleonora Russo, and Andrea Spallarossa. 2021. "Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective" International Journal of Molecular Sciences 22, no. 14: 7641. https://doi.org/10.3390/ijms22147641
APA StyleBrullo, C., Villa, C., Tasso, B., Russo, E., & Spallarossa, A. (2021). Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. International Journal of Molecular Sciences, 22(14), 7641. https://doi.org/10.3390/ijms22147641