
The 3,4-Quinones of Estrone and Estradiol Are the Initiators of Cancer whereas Resveratrol and N-acetylcysteine Are the Preventers

Abstract
:1. Introduction
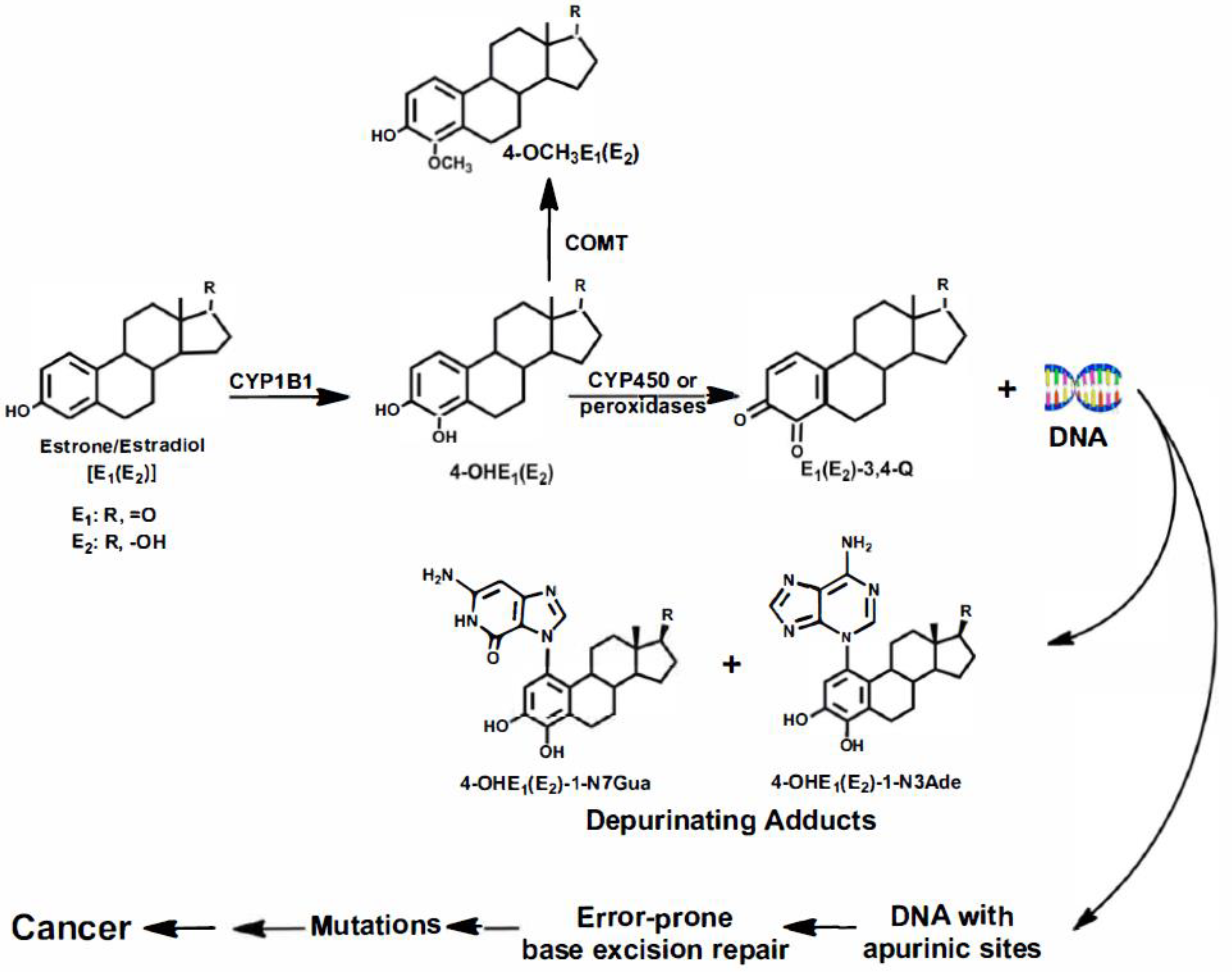
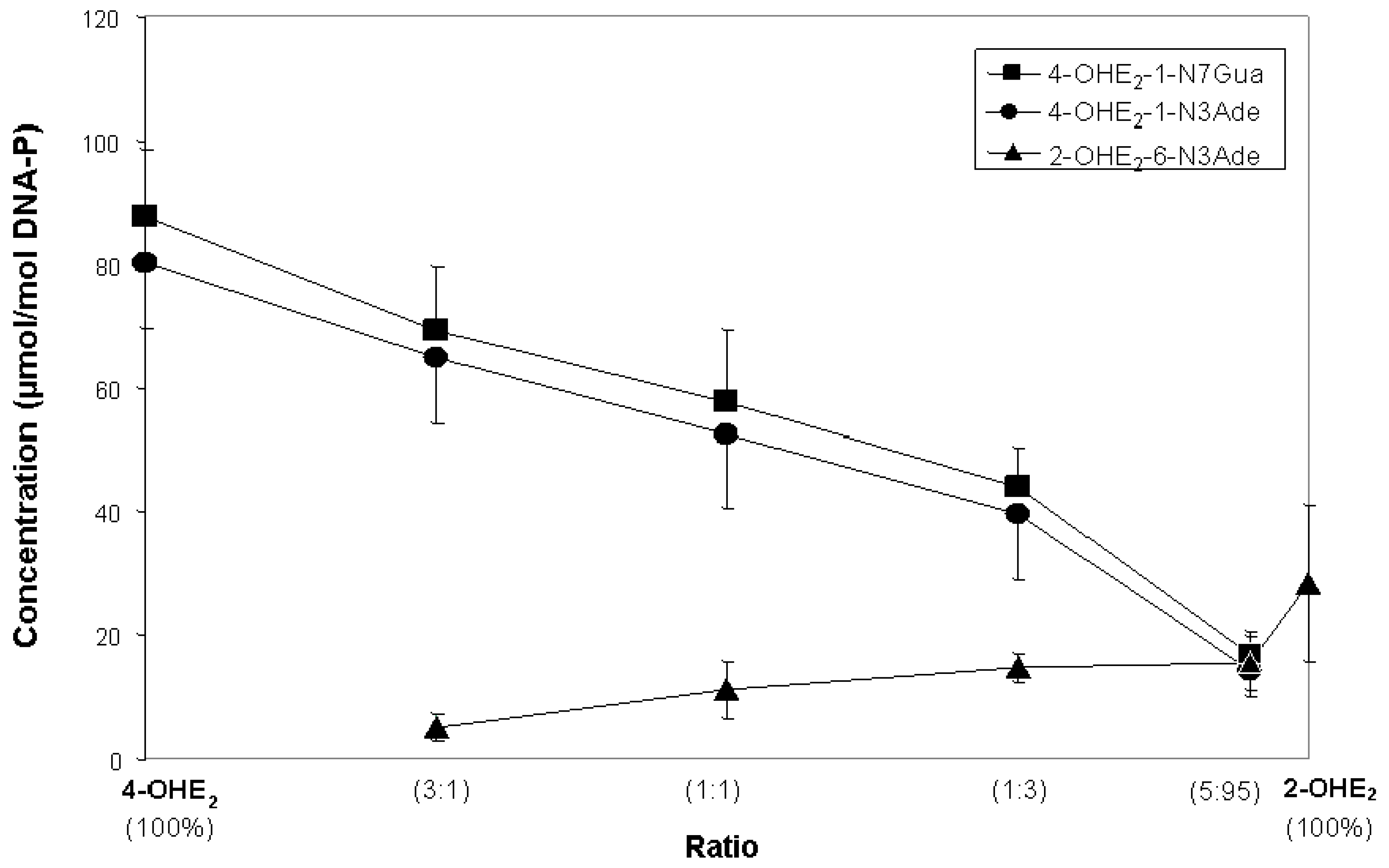
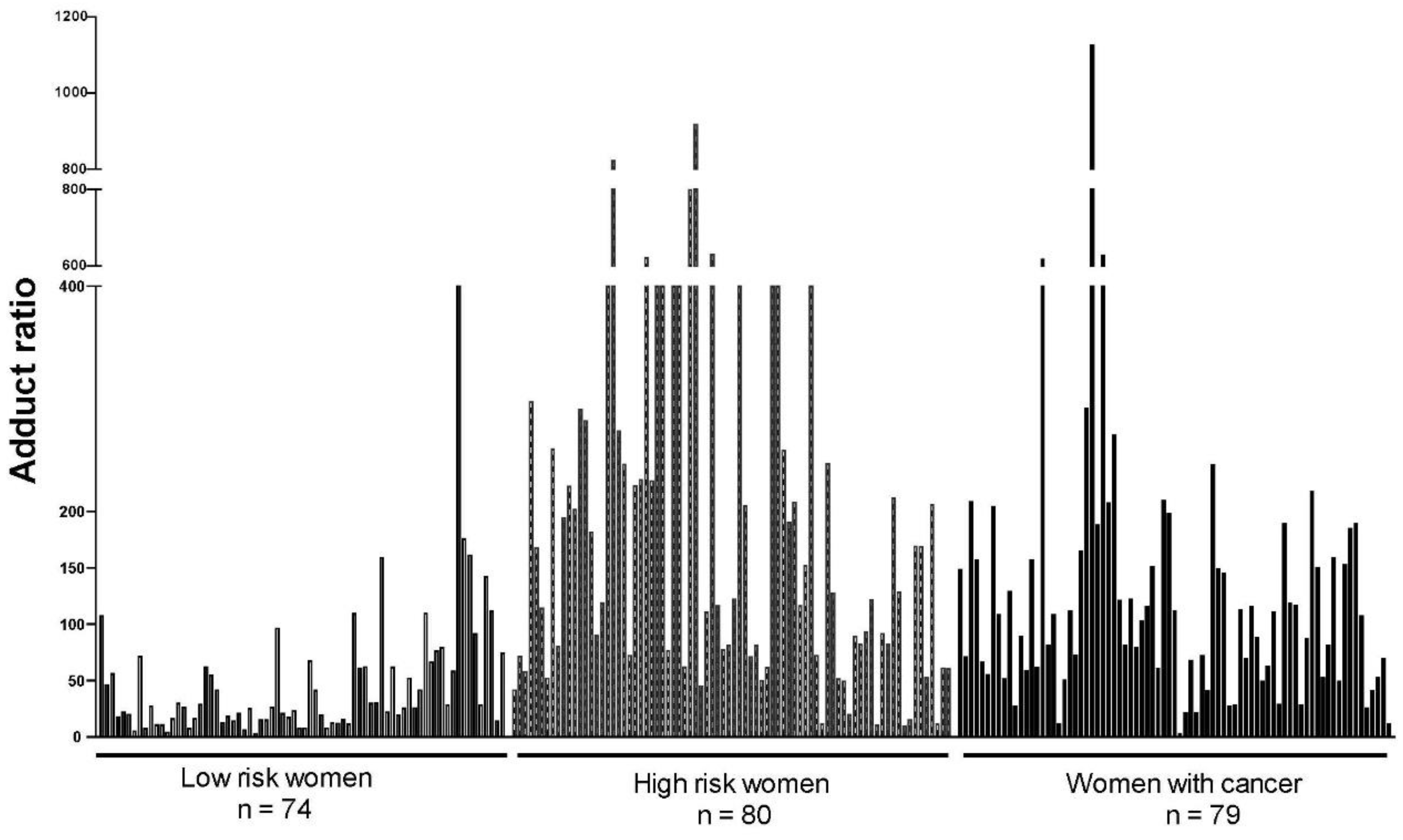
2. Initiation of Cancer
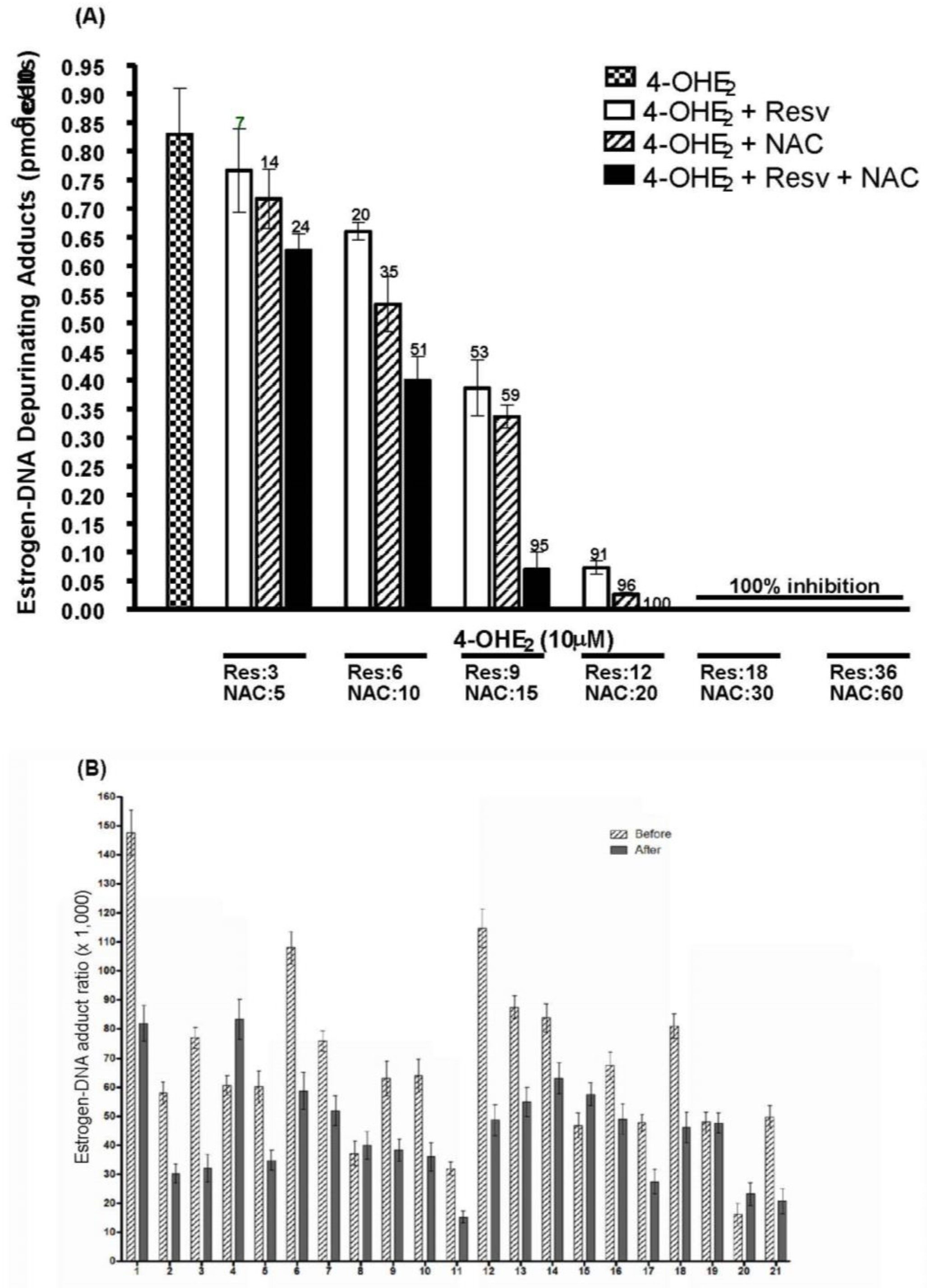
3. Prevention of Cancer by Inhibition of Estrogen-DNA Adduct Formation
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, S.B.; Hankinson, S.E. Endogenous estrogens and the risk of breast, endometrial, and ovarian cancers. Steroids 2015, 99, 8–10. [Google Scholar] [CrossRef]
- Yager, J.D. Mechanisms of estrogen carcinogenesis: The role of E2/E1-quinone metabolites suggests new approaches to preventive intervention—A review. Steroids 2015, 99, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Bosland, M.C.; Ford, H.; Horton, L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17β or diethylstilbestrol. Carcinogenesis 1995, 16, 1311–1317. [Google Scholar] [CrossRef]
- Ho, S.-M.; Lee, M.-T.; Lam, H.-M.; Keung, Y.-K. Estrogens and prostate cancer: Etiology, mediators, prevention, and management. Endocrinol. Metab. Clin. N. Am. 2011, 40, 591–614. [Google Scholar] [CrossRef] [Green Version]
- Rohrmann, S.; Nelson, W.G.; Rifari, N.; Brown, T.R.; Dobs, A.; Kanarek, N.; Yager, J.D.; Platz, E.A. Serum estrogen, but not testosterone, levels differ between black and white men in a nationally representative sample of Americans. J. Clin. Endocrinol. Metab. 2007, 92, 2519–2525. [Google Scholar] [CrossRef]
- Ross, R.; Bernstein, L.; Judd, H.; Hanisch, R.; Pike, M.; Henderson, B. Serum testosterone levels in healthy young black and white men. J. Natl. Cancer Inst. 1986, 76, 45–58. [Google Scholar]
- Orwoll, E.S.; Nielson, C.M.; Labrie, F.; Barrett-Connor, E.; Cauley, J.A.; Cummings, S.R.; Ensrud, K.; Karlsson, M.; Lau, E.; Leung, P.C.; et al. Evidence of geographical and racial variation in serum sex steroid levels in older men. J. Clin. Endocrinol. Metab. 2010, 95, E151–E160. [Google Scholar] [CrossRef]
- Krieg, M.; Nass, R.; Runn, S. Effect of aging on endogenous level of 5 alpha-dihydrotestosterone, testosterone, estradiol, and estrone in epithelium and stroma of normal and hyperplastic human prostate. J. Clin. Endocrinol. Metab. 1993, 77, 375–381. [Google Scholar] [PubMed] [Green Version]
- McPherson, S.J.; Wang, H.; Jones, M.E.; Pedersen, J.; Iismaa, T.O.; Wreford, N.; Simpson, E.R.; Risbridger, G.P. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology 2001, 142, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, E.L.; Kumar, S.; Todorovic, R.; Higginbotham, S.; Badawi, A.F.; Rogan, E.G. Imbalance of estrogen homeostasis in kidney and liver of hamsters treated with estradiol: Implications for estrogen-induced initiation of renal tumors. Chem. Res. Toxicol. 2001, 14, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Rogan, E.G.; Badawi, A.F.; Devanesan, P.D.; Meza, J.L.; Edney, J.A.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, E.; Rogan, E. The molecular etiology and prevention of estrogen-initiated cancers. Mol. Asp. Med. 2014, 36, 1–55. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, E.L.; Rogan, E.G.; Zahid, M. Critical depurinating DNA adducts: Estrogen adducts in the etiology and prevention of cancer and dopamine adducts in the etiology and prevention of Parkinson’s disease. Int. J. Cancer 2017, 141, 1078–1090. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, D.; Pelling, J.C.; Cavalieri, E.L.; Rogan, E.G. Relating aromatic hydrocarbon-induced DNA adducts and c-Harvey-ras mutations in mouse skin papillomas: The role of apurinic sites. Proc. Natl. Acad. Sci. USA 1995, 92, 10422–10426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, D.; Mailander, P.; Li, K.M.; Higginbotham, S.; Zhang, H.L.; Gross, M.L.; Cavalieri, E.; Rogan, E. Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene 2001, 20, 7945–7953. [Google Scholar] [CrossRef] [Green Version]
- Mailander, P.C.; Meza, J.L.; Higginbotham, S.; Chakravarti, D. Induction of A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J. Steroid. Biochem. Mol. Biol. 2006, 101, 204–215. [Google Scholar] [CrossRef]
- Zhu, B.T.; Conney, A.H. Functional role of estrogen metabolism in target cells: Review and perspectives. Carcinogenesis 1998, 9, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalieri, E.L.; Devanesan, P.; Bosland, M.C.; Badawi, A.F.; Rogan, E.G. Catechol estrogen metabolites and conjugates in different regions of the prostate of Noble rats treated with 4-hydroxyestradiol: Implications for estrogen-induced initiation of prostate cancer. Carcinogenesis 2002, 23, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Devanesan, P.; Santen, R.J.; Bocchinfuso, W.P.; Korach, K.S.; Rogan, E.G.; Cavalieri, E.L. Catechol estrogen metabolites and conjugates in mammary tumors and hyperplastic tissue from estrogen receptor alpha knock out (ERKO)/Wnt 1 mice; implications for initiation of mammary tumors. Carcinogenesis 2001, 22, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Bocchinfuso, W.P.; Korach, K.S. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J. Mammary Gland Biol. Neoplasia 1997, 2, 323–334. [Google Scholar] [CrossRef]
- Bocchinfuso, W.P.; Hively, W.P.; Couse, J.F.; Varmus, H.E.; Korach, K.S. A mouse mammary tumor virus-Wnt-1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor-α. Cancer Res. 1999, 59, 1869–1876. [Google Scholar] [PubMed]
- Santen, R.J.; Yue, W.; Bocchinfuso, W.; Korach, K.; Wang, J.P.; Rogan, E.G.; Yang, L.; Cavalieri, E.; Russo, J.; Devanesan, P.; et al. Estradiol-induced carcinogenesis via formation of genotoxic metabolites. In Advances in Endocrine Therapy of Breast Cancer; Ingle, J.N., Dowsett, M., Eds.; Marcel Dekker: New York, NY, USA, 2003; pp. 163–177. [Google Scholar]
- Yue, W.; Santen, R.J.; Wang, J.P.; Li, Y.; Verderame, M.F.; Bocchinfuso, W.P.; Korach, K.S.; Todorovic, R.; Rogan, E.G.; Cavalieri, E.L. Genotoxic metabolites of estradiol in breast: Potential mechanism of estradiol induced carcinogenesis. J. Steroid Biochem. Mol. Biol. 2003, 86, 477–486. [Google Scholar] [CrossRef]
- Santen, R.; Cavalieri, E.; Rogan, E.; Russo, J.; Guttenplan, J.; Ingle, J.; Yue, W. Estrogen mediation of breast tumor formation involves estrogen receptor-dependent, as well as independent, genotoxic effects. Ann. N. Y. Acad. Sci. 2009, 1155, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Lareef, M.H.; Tahin, Q.; Hu, Y.F.; Slater, C.; Ao, X.; Russo, I.H. 17-Beta-estradiol is carcinogenic in human breast epithelial cells. J. Steroid Biochem. Mol. Biol. 2002, 80, 149–162. [Google Scholar] [CrossRef]
- Russo, J.; Hasan Lareef, M.; Balogh, G.; Guo, S.; Russo, I.H. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J. Steroid Biochem. Mol. Biol. 2003, 87, 1–25. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. Genotoxicity of steroidal estrogens. Trends Endocrinol. Metab. 2004, 15, 211–215. [Google Scholar] [CrossRef]
- Russo, J.; Fernandez, S.V.; Russo, P.A.; Fernbaugh, R.; Sheriff, F.S.; Lareef, H.M.; Garber, J.; Russo, I.H. 17β-Estradiol induces transformations and tumorigenesis in human breast epithelial cells. FASEB J. 2006, 20, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Saeed, M.; Beseler, C.; Rogan, E.G.; Cavalieri, E.L. Resveratrol and N-acetylcysteine block the cancer-initiating step in MCF-10F cells. Free Radic. Biol. Med. 2011, 50, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Zahid, M.; Wang, C.; Saeed, M.; Cavalieri, E.L.; Rogan, E.G. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev. Res. 2008, 1, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.; Gaikwad, N.W.; Ali, M.F.; Lu, F.; Saeed, M.; Yang, L.; Rogan, E.G.; Cavalieri, E.L. Prevention of estrogen-DNA adducts in MCF-10F cells by resveratrol. Free Radic. Biol. Med. 2008, 45, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Saeed, M.; Ali, M.F.; Rogan, E.G.; Cavalieri, E.L. N-Acetylcysteine blocks formation of cancer-initiating estrogen-DNA adducts in cells. Free Radic. Biol. Med. 2010, 49, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Lareef, M.H.; Garber, J.; Russo, P.A.; Russo, I.H.; Heulings, R.; Russo, J. The estrogen antagonist ICI-182-780 does not inhibit the transformation phenotypes induced by 17-beta-estradiol and 4-OH estradiol in human breast epithelial cells. Int. J. Oncol. 2005, 26, 423–429. [Google Scholar] [PubMed]
- Stack, D.E.; Byun, J.; Gross, M.L.; Rogan, E.G.; Cavalieri, E.L. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem. Res. Toxicol. 1996, 9, 851–859. [Google Scholar] [CrossRef]
- Li, K.M.; Todorovic, R.; Devanesan, P.; Higginbotham, S.; Kofeler, H.; Ramanathan, R.; Gross, M.L.; Rogan, E.G.; Cavalieri, E.L. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis 2004, 25, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.; Kohli, E.; Saeed, M.; Rogan, E.; Cavalieri, E. The greater reactivity of estradiol-3,4-quinone versus estradiol-2,3-quinone with DNA in the formation of depurinating adducts. Implications for tumor-initiating activity. Chem. Res. Toxicol. 2006, 19, 164–172. [Google Scholar] [CrossRef]
- Liehr, J.G.; Fang, W.F.; Sirbasku, D.A.; Ari-Ulubelen, A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem. 1986, 24, 353–356. [Google Scholar] [CrossRef]
- Li, J.J.; Li, S.A. Estrogen carcinogenesis in Syrian hamster tissues: Role of metabolism. Fed. Proc. 1987, 46, 1858–1863. [Google Scholar]
- Newbold, R.R.; Liehr, J.G. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000, 60, 235–237. [Google Scholar]
- Cavalieri, E. Minisymposium on endogenous carcinogens: The catechol estrogen pathway. An introduction. Polycycl. Aromat. Compd. 1994, 6, 223–228. [Google Scholar] [CrossRef]
- Mobley, J.A.; Bhat, A.S.; Brueggemeier, R.W. Measurement of oxidative DNA damage by catechol estrogens and analogues in vitro. Chem. Res. Toxicol. 1999, 12, 270–277. [Google Scholar] [CrossRef]
- Singh, B.; Mense, S.M.; Remotti, F.; Liu, X.; Bhat, H.K. Antioxidant butylated hydroxyanisole inhibits estrogen-induced breast carcinogenesis in female ACI rats. J. Biochem. Mol. Toxicol. 2009, 23, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, N.W.; Yang, L.; Muti, P.; Meza, J.L.; Pruthi, S.; Ingle, J.N.; Rogan, E.G.; Cavalieri, E.L. The molecular etiology of breast cancer: Evidence from biomarkers of risk. Int. J. Cancer 2008, 122, 1949–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaikwad, N.W.; Yang, L.; Pruthi, S.; Ingle, J.N.; Sandhu, N.; Rogan, E.; Cavalieri, E.L. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer Basic Clin. Res. 2009, 3, BCBCR-S2112. [Google Scholar] [CrossRef] [PubMed]
- Pruthi, S.; Yang, L.; Sandhu, N.P.; Ingle, J.N.; Beseler, C.L.; Suman, V.J.; Cavalieri, E.L.; Rogan, E.G. Evaluation of serum estrogen-DNA adducts as potential biomarkers of breast cancer risk. J. Steroid Biochem. Mol. Biol. 2012, 132, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.; Mondal, B.; LeVan, T.D.; Rogan, E.G. Estrogen metabolism in African-American women with and without breast cancer: A pilot study. Chem. Res. Toxicol. 2018, 32, 190–194. [Google Scholar] [CrossRef]
- Zahid, M.; Goldner, W.; Beseler, C.L.; Rogan, E.G.; Cavalieri, E.L. Unbalanced estrogen metabolism in thyroid cancer. Int. J. Cancer 2013, 133, 2642–2649. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.; Beseler, C.L.; Hall, J.B.; LeVan, T.; Cavalieri, E.L.; Rogan, E.G. Unbalanced estrogen metabolism in ovarian cancer. Int. J. Cancer 2014, 134, 2414–2423. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Gaikwad, N.; Meza, J.; Cavalieri, E.; Muti, P.; Trock, B.; Rogan, E. Novel biomarkers for risk of prostate cancer: Results from a case-control study. Prostate 2009, 69, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaikwad, N.W.; Yang, L.; Weisenburger, D.D.; Vose, J.; Beseler, C.; Rogan, E.G.; Cavalieri, E. Urinary biomarkers suggest that estrogen-DNA adducts may play a role in the aetiology of non-Hodgkin lymphoma. Biomarker 2009, 14, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Markushin, Y.; Gaikwad, N.; Zhang, H.; Rogan, E.; Cavalieri, E.; Trock, B.; Pavlovich, C.; Jankowiak, R. Potential biomarker for early risk assessment of prostate cancer. Prostate 2006, 66, 1565–1571. [Google Scholar] [CrossRef]
- Zahid, M.; Gaikwad, N.; Rogan, E.G.; Cavalieri, E.L. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem. Res. Toxicol. 2007, 20, 1947–1953. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Mohannadi, A.N.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential adverse effects of resveratrol: A literature review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, D.; Zahid, M.; Mailander, P.C.; Meza, J.L.; Rogan, E.G.; Cavalieri, E.L.; Chakravarti, D. Reduction of estrogen-induced transformation of mouse mammary epithelial cells by N-acetylcysteine. J. Steroid Biochem. Mol. Biol. 2008, 109, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muran, P.; Muran, S.; Beseler, C.L.; Cavalieri, E.L.; Rogan, E.G.; Zahid, M. Breast health and reducing breast cancer risk, a functional medicine approach. J. Altern. Complement. Med. 2015, 21, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Preston-Martin, S.; Pike, M.C.; Ross, R.K.; Jones, P.A.; Henderson, B.E. Increased cell division as a cause of human cancer. Cancer Res. 1990, 50, 7415–7421. [Google Scholar]
- Cuzick, J. International-breast-cancer-intervention-study. A brief review of the international breast cancer intervention study (IBIS), the other current breast cancer prevention trials, and proposals for future trials. Ann. N. Y. Acad. Sci. 2001, 949, 123–133. [Google Scholar] [CrossRef]
- Cummings, S.R.; Eckert, S.; Krueger, K.A.; Grady, D.; Powles, T.G.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. Multiple outcomes of raloxifene evaluation. JAMA 1999, 281, 2189–2197. [Google Scholar] [CrossRef] [Green Version]
- Martino, S.; Cauley, J.A.; Barrett-Connor, E.; Powles, T.J.; Mershon, J.; Disch, D.; Secrest, R.J.; Cummings, S.R. For the CORE Investigators. Continuing outcomes relevant to Evista: Breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J. Natl. Cancer Inst. 2004, 96, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2020; p. 4. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalieri, E.; Rogan, E. The 3,4-Quinones of Estrone and Estradiol Are the Initiators of Cancer whereas Resveratrol and N-acetylcysteine Are the Preventers. Int. J. Mol. Sci. 2021, 22, 8238. https://doi.org/10.3390/ijms22158238
Cavalieri E, Rogan E. The 3,4-Quinones of Estrone and Estradiol Are the Initiators of Cancer whereas Resveratrol and N-acetylcysteine Are the Preventers. International Journal of Molecular Sciences. 2021; 22(15):8238. https://doi.org/10.3390/ijms22158238
Chicago/Turabian StyleCavalieri, Ercole, and Eleanor Rogan. 2021. "The 3,4-Quinones of Estrone and Estradiol Are the Initiators of Cancer whereas Resveratrol and N-acetylcysteine Are the Preventers" International Journal of Molecular Sciences 22, no. 15: 8238. https://doi.org/10.3390/ijms22158238
APA StyleCavalieri, E., & Rogan, E. (2021). The 3,4-Quinones of Estrone and Estradiol Are the Initiators of Cancer whereas Resveratrol and N-acetylcysteine Are the Preventers. International Journal of Molecular Sciences, 22(15), 8238. https://doi.org/10.3390/ijms22158238