
Investigation of the Complexes Formed between PARP1 Inhibitors and PARP1 G-Quadruplex at the Gene Promoter Region

 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
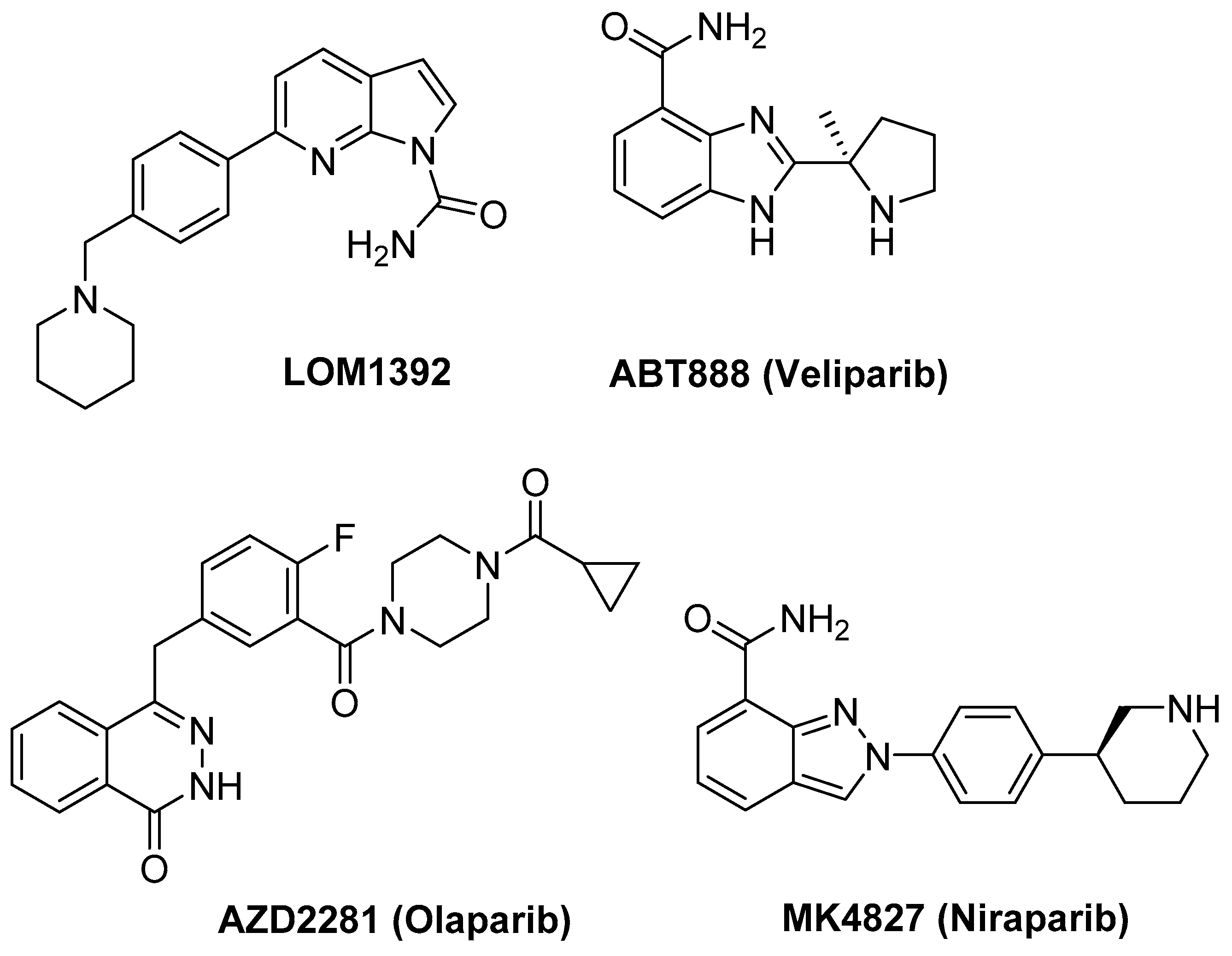
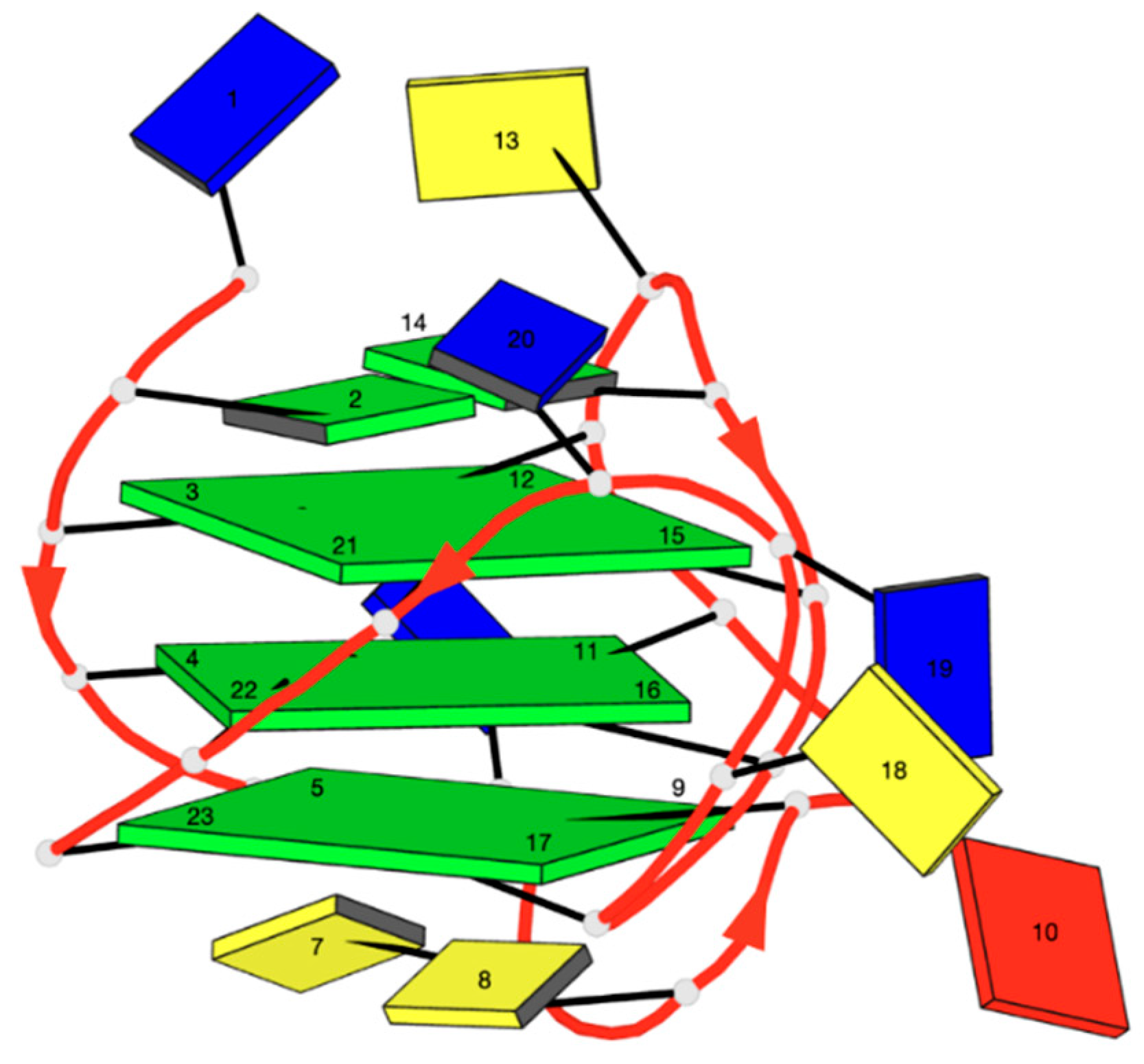
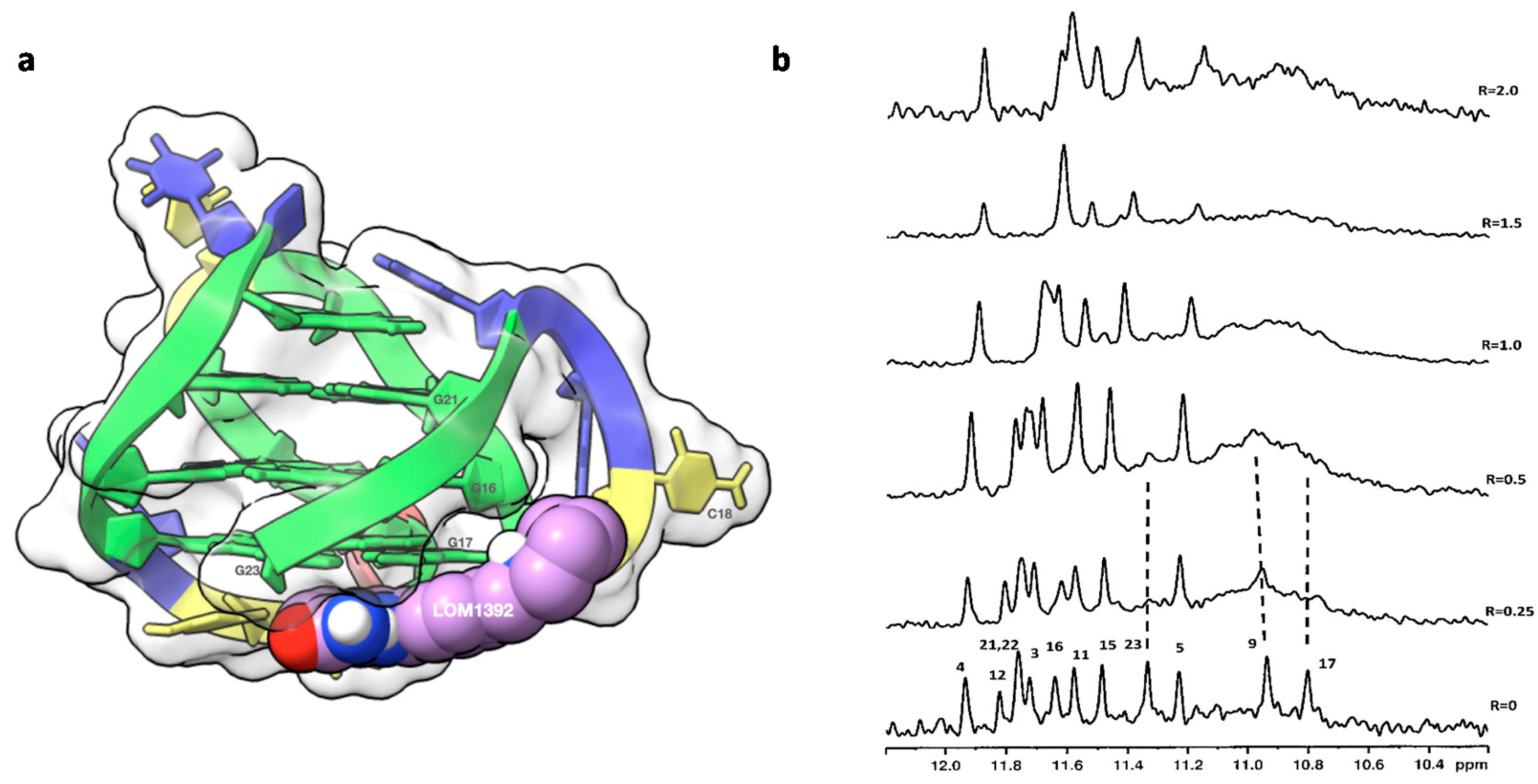
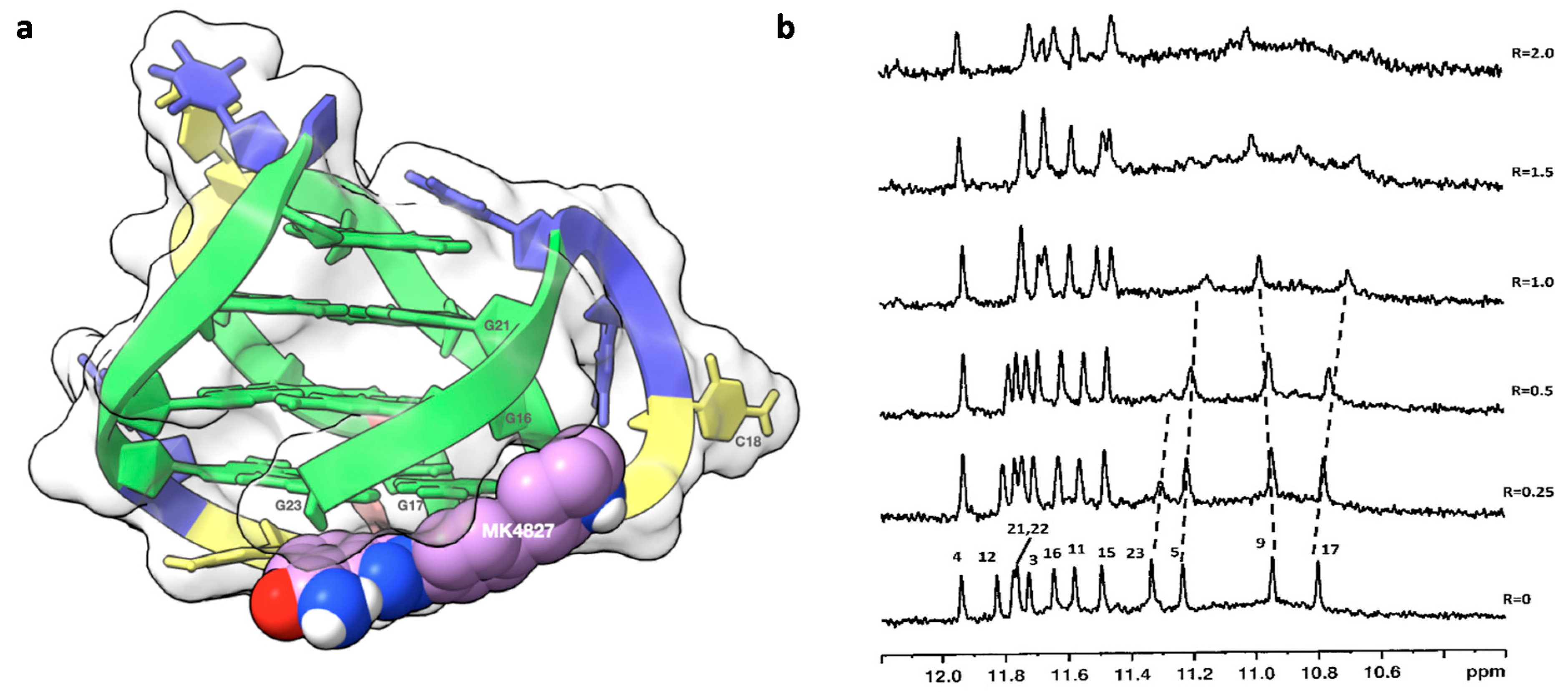
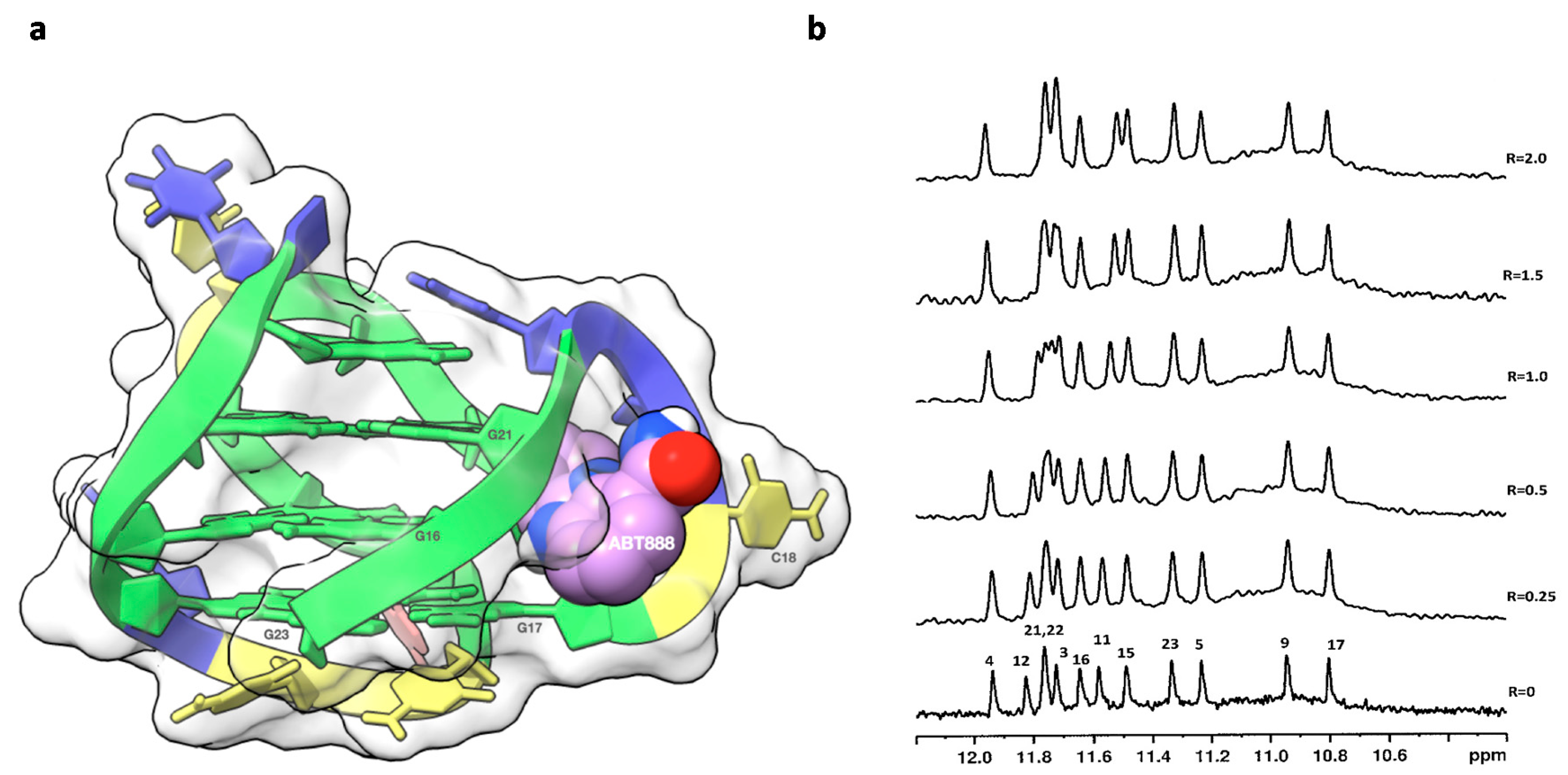
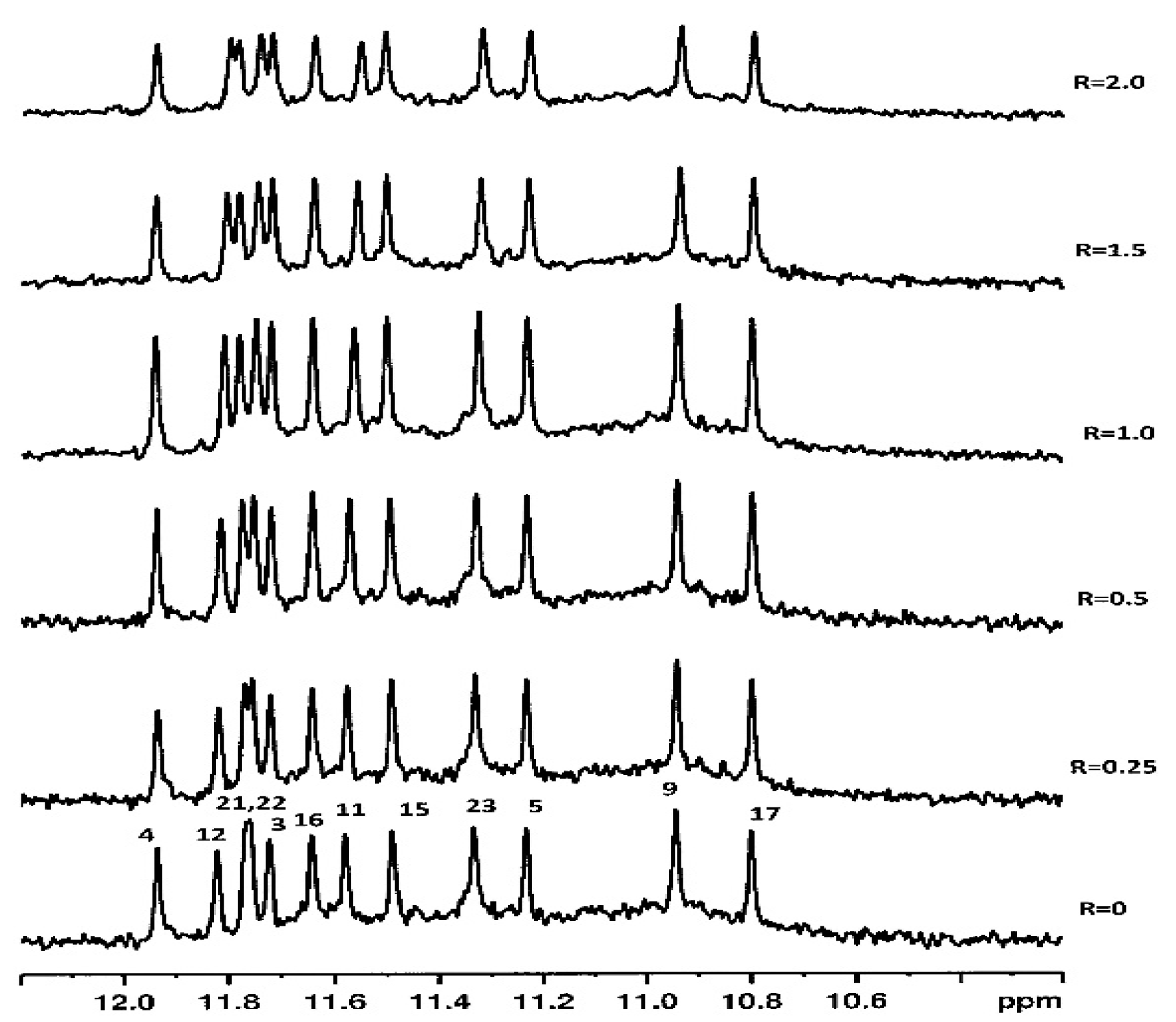
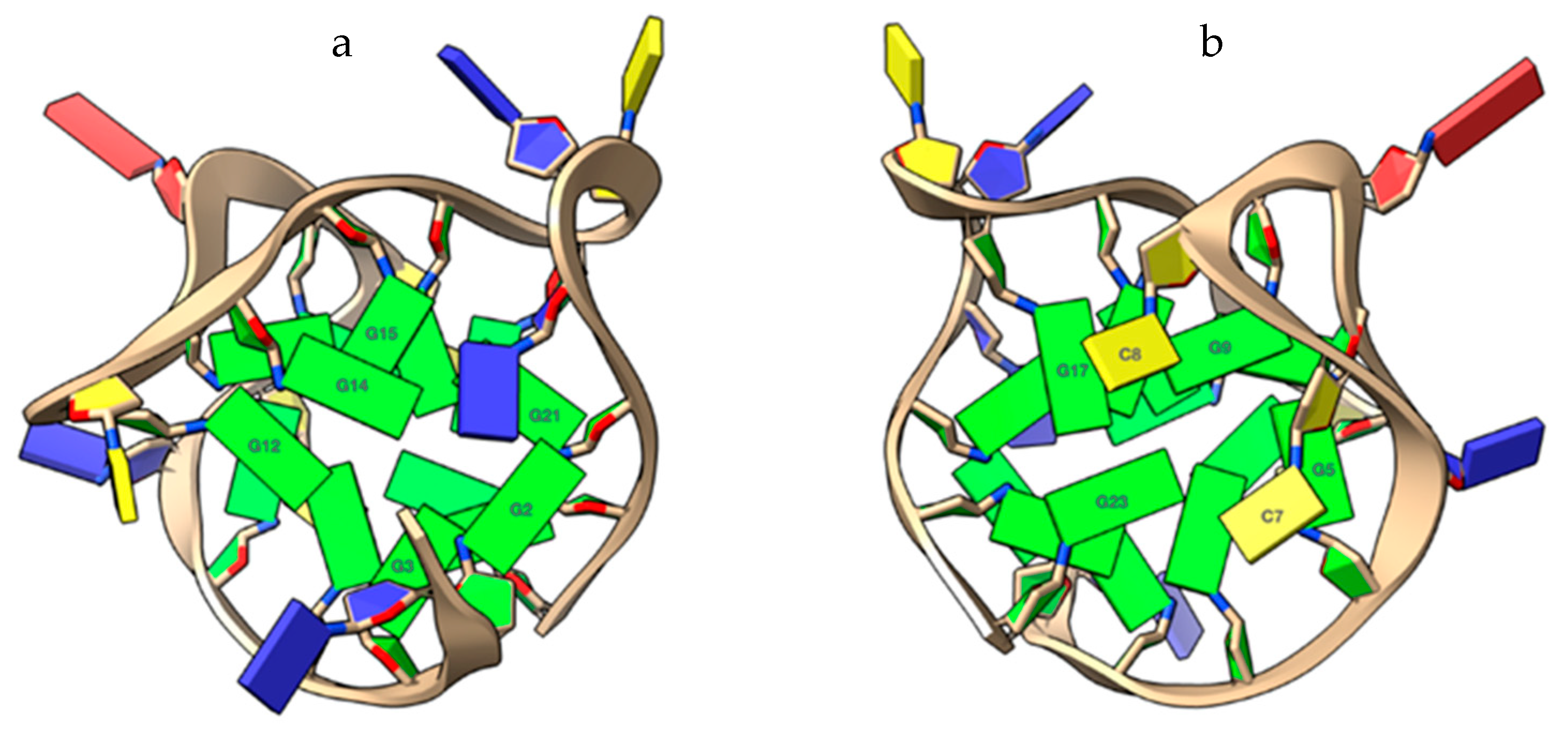
2.1. 1H NMR Experiments and Molecular Modeling on the LOM1392, ABT888, MK4827, and AZD2281 Complexes with TP3-T6 (5′-D TGGGGT6CCGAGGCGGGGCTTGGG-3′)
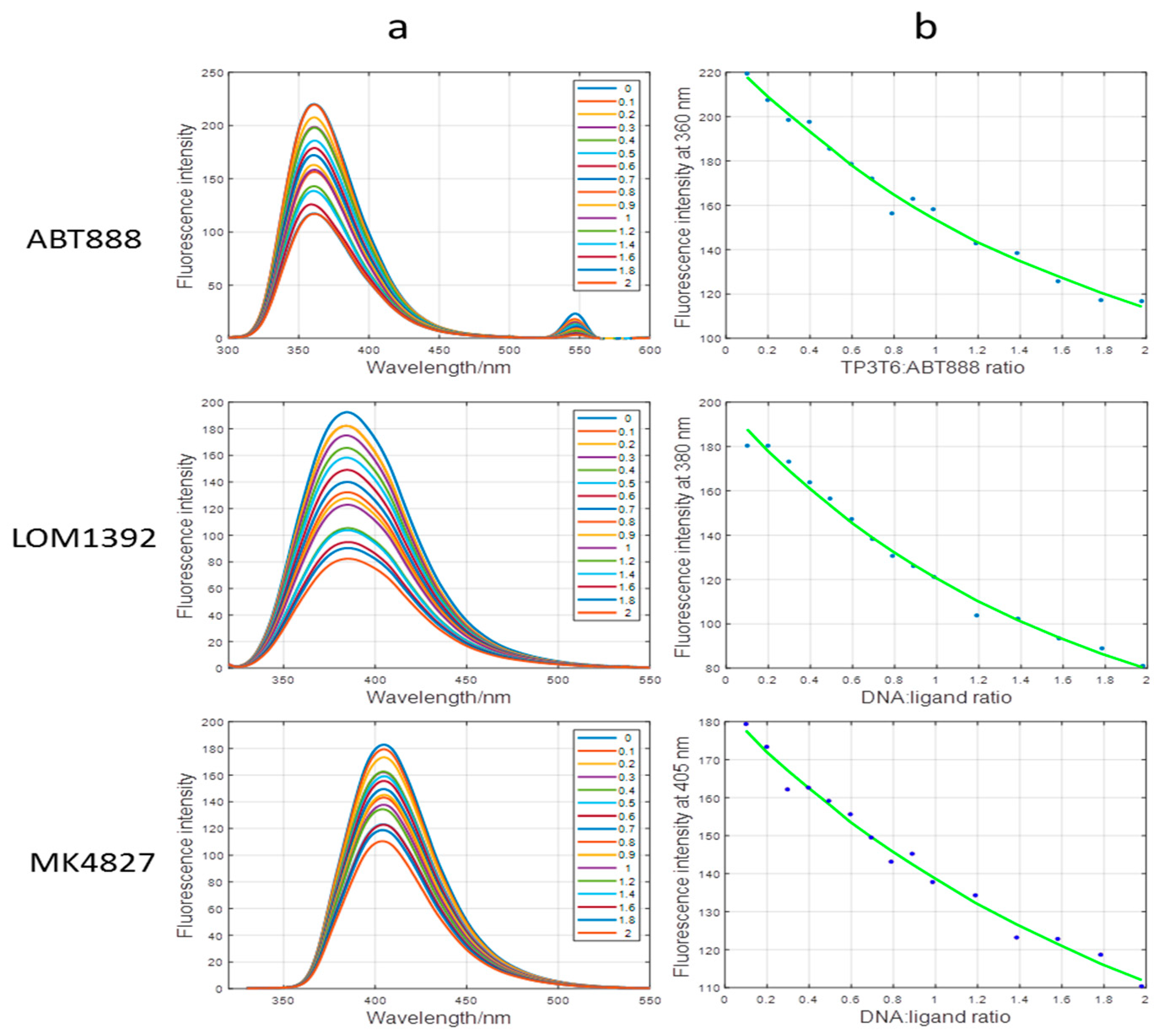
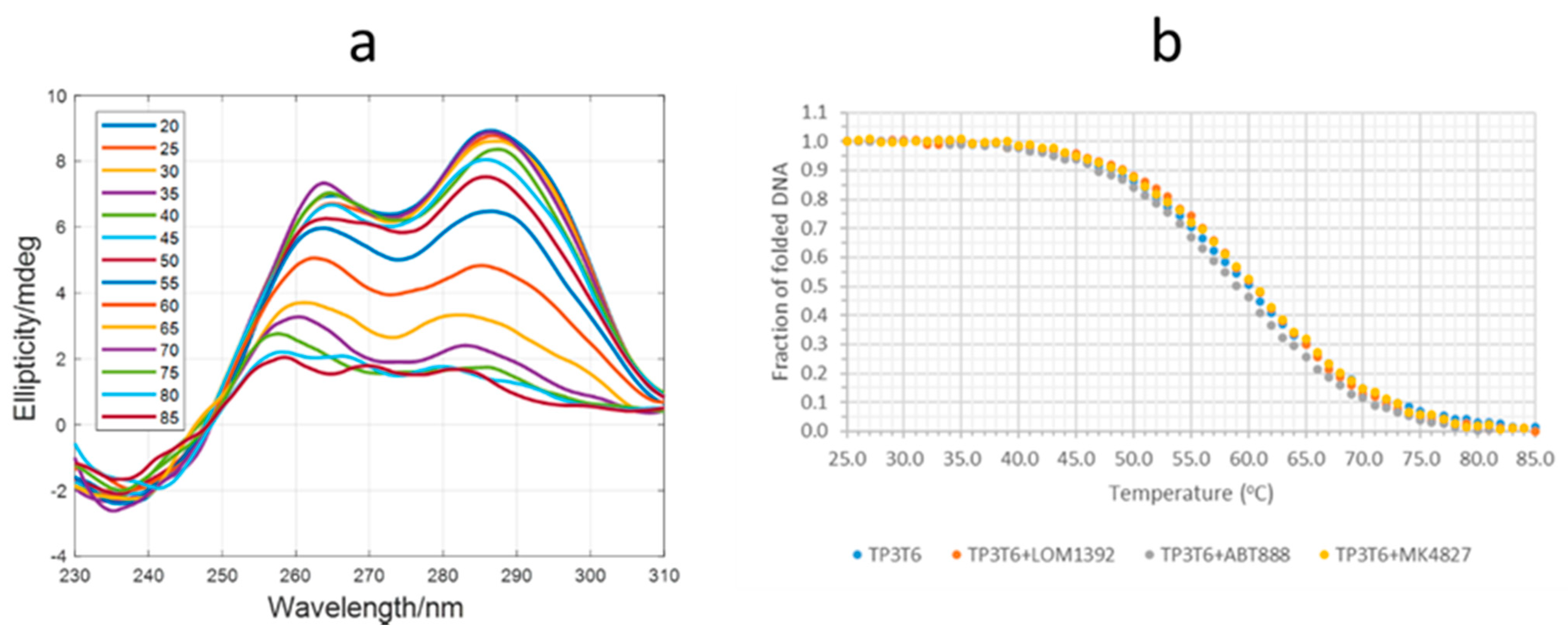
2.2. Fluorescence and CD Studies on the LOM1392, ABT888, and MK4827 Complexes with TP3-T6 (5′-D TGGGGT6CCGAGGCGGGGCTTGGG-3′)
3. Materials and Methods
3.1. Ligands
3.2. Sample Preparation
3.3. NMR Experiments
3.4. CD and Fluorescence
3.5. Molecular Modeling Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.R.; Nussenzweig, A.R.C.A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.-X.; Jiang, T.; Long, J.; Ma, Z.-Y.; Lu, A.-P.; Cheng, Y.; Cao, D.-S. The ups and downs of Poly(ADP-ribose) Polymerase-1 inhibitors in cancer therapy–Current progress and future direction. Eur. J. Med. Chem. 2020, 203, 112570. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Cincinelli, R.; Musso, L.; Merlini, L.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Carenini, N.; Perego, P.; Penco, S.; Artali, R.; et al. 7-Azaindole-1-carboxamides as a new class of PARP-1 inhibitors. Bioorg. Med. Chem. 2014, 22, 1089–1103. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Sengupta, A.; Ganguly, A.; Chowdhury, S. Promise of G-Quadruplex Structure Binding Ligands as Epigenetic Modifiers with Anti-Cancer Effects. Molecules 2019, 24, 582. [Google Scholar] [CrossRef] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengar, A.; Vandana, J.J.; Chambers, V.S.; Di Antonio, M.; Winnerdy, F.R.; Balasubramanian, S.; Phan, A.T. Structure of a (3 + 1) hybrid G-quadruplex in the PARP1 promoter. Nucleic Acids Res. 2019, 47, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Dallavalle, S.; Musso, L.; Artali, R.; Aviñó, A.; Scaglioni, L.; Eritja, R.; Gargallo, R.; Mazzini, S. G-quadruplex binding properties of a potent PARP-1 inhibitor derived from 7-azaindole-1-carboxamide. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2017, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Dyson, R.M.; Kaderli, S.; Lawrance, G.A.; Maeder, M. Second order global analysis: The evaluation of series of spectrophotometric titrations for improved determination of equilibrium constants. Anal. Chim. Acta 1997, 353, 381–393. [Google Scholar] [CrossRef]
- Gargallo, R. Hard/Soft hybrid modeling of temperature-induced unfolding processes involving G-quadruplex and i-motif nucleic acid structures. Anal. Biochem. 2014, 466, 4–15. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a La-marckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Perez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoureux, G.; Roux, B. Modeling induced polarization with classical Drude oscillators: Theory and molecular dynamics simulation algorithm. J. Chem. Phys. 2003, 119, 3025–3039. [Google Scholar] [CrossRef]
- Jiang, W.; Hardy, D.J.; Phillips, J.C.; MacKerell, A.D., Jr.; Schulten, K.; Roux, B. High-performance scalable molecular dynamics simulations of a polarizable force field based on classical Drude oscillators in NAMD. J. Phys. Chem. Lett. 2011, 2, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- A Darden, T.; York, D.M.; Pedersen, L.G. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.-J. DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL. Nucleic Acids Res. 2020, 48, e74. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Tm (°C) | ΔH (kcal·mol−1) | ΔS (cal·K−1·mol−1) | ΔG37 °C (kcal·mol−1) |
|---|---|---|---|---|
| TP3T6 | 59.6 ± 0.8 | −39.0 ± 0.4 | −117.2 ± 1.0 | −2.7 ± 1.1 |
| TP3T6:LOM1392 | 60.2 ± 1.0 | −43.1 ± 0.5 | −129.2 ± 1.5 | −3.0 ± 1.6 |
| TP3T6:ABT888 | 58.8 ± 1.3 | −41.1 ± 0.6 | −123.7 ± 1.9 | −2.7 ± 2.0 |
| TP3T6:MK4827 | 60.3 ± 1.3 | −41.1 ± 0.6 | −123.0 ± 1.8 | −3.0 ± 1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallavalle, S.; Princiotto, S.; Mattio, L.M.; Artali, R.; Musso, L.; Aviñó, A.; Eritja, R.; Pisano, C.; Gargallo, R.; Mazzini, S. Investigation of the Complexes Formed between PARP1 Inhibitors and PARP1 G-Quadruplex at the Gene Promoter Region. Int. J. Mol. Sci. 2021, 22, 8737. https://doi.org/10.3390/ijms22168737
Dallavalle S, Princiotto S, Mattio LM, Artali R, Musso L, Aviñó A, Eritja R, Pisano C, Gargallo R, Mazzini S. Investigation of the Complexes Formed between PARP1 Inhibitors and PARP1 G-Quadruplex at the Gene Promoter Region. International Journal of Molecular Sciences. 2021; 22(16):8737. https://doi.org/10.3390/ijms22168737
Chicago/Turabian StyleDallavalle, Sabrina, Salvatore Princiotto, Luce M. Mattio, Roberto Artali, Loana Musso, Anna Aviñó, Ramon Eritja, Claudio Pisano, Raimundo Gargallo, and Stefania Mazzini. 2021. "Investigation of the Complexes Formed between PARP1 Inhibitors and PARP1 G-Quadruplex at the Gene Promoter Region" International Journal of Molecular Sciences 22, no. 16: 8737. https://doi.org/10.3390/ijms22168737
APA StyleDallavalle, S., Princiotto, S., Mattio, L. M., Artali, R., Musso, L., Aviñó, A., Eritja, R., Pisano, C., Gargallo, R., & Mazzini, S. (2021). Investigation of the Complexes Formed between PARP1 Inhibitors and PARP1 G-Quadruplex at the Gene Promoter Region. International Journal of Molecular Sciences, 22(16), 8737. https://doi.org/10.3390/ijms22168737