
Aflatoxin Exposure during Early Life Is Associated with Differential DNA Methylation in Two-Year-Old Gambian Children

 , ,
, ,
Abstract
:1. Introduction
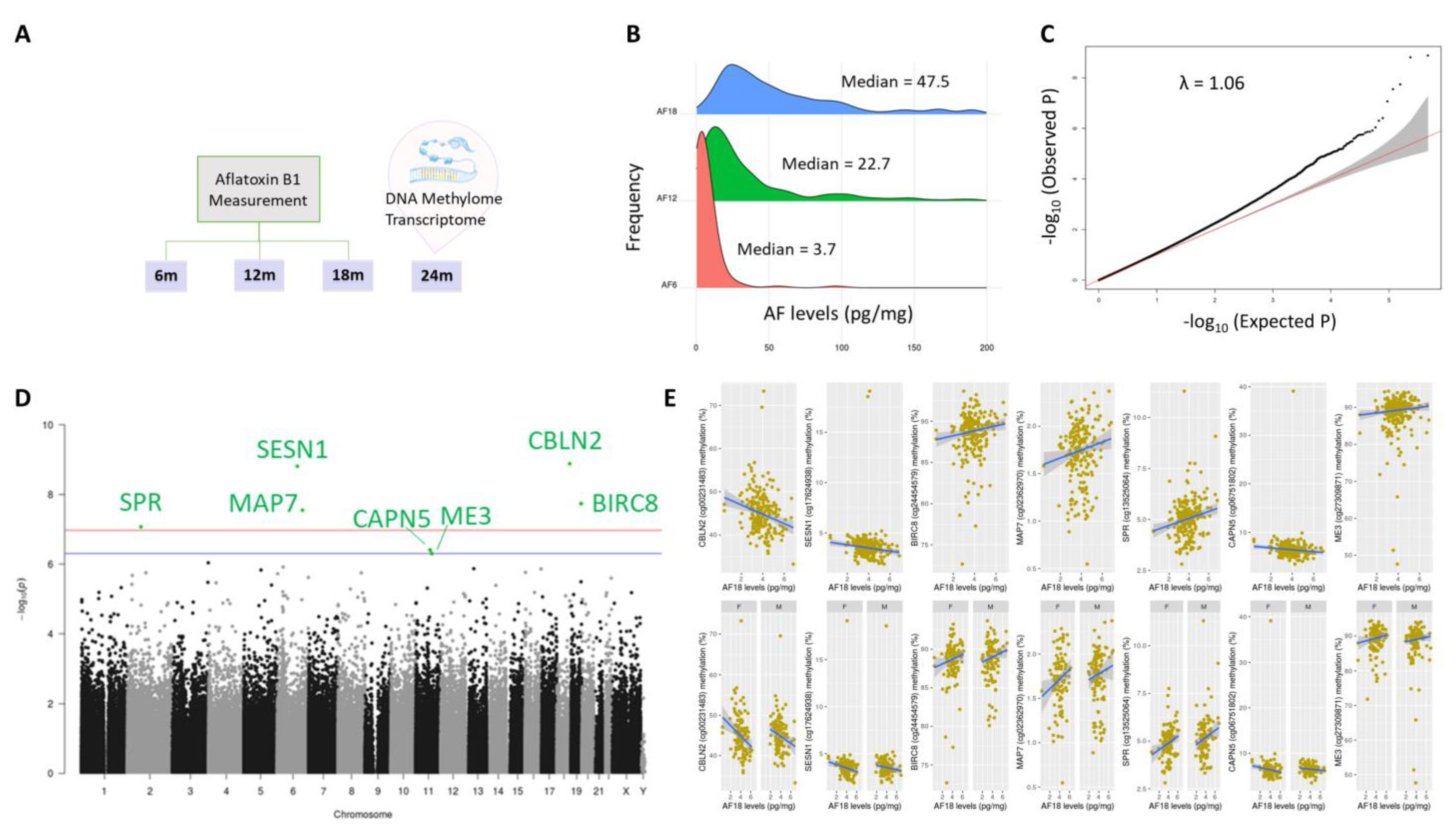
2. Results
2.1. Aflatoxin Exposure over Time in Early Life
2.2. DNA Methylation Markers of Aflatoxin Exposure and Associated Biological Pathways: Continuous Model
2.3. DNA Methylation Markers of Aflatoxin Exposure and Associated Biological Pathways: Categorical Time-Dose Model
2.4. Genomic Distribution and Functional Characterization of Identified DNA Methylation Markers of Aflatoxin Exposure
3. Methods
3.1. Study Population and Blood Sampling
3.2. AF-alb Analysis
3.3. Bisulfite Conversion
3.4. Genome-Wide Methylation Array
3.5. DNA Methylome Data Pre-Processing
3.6. Assessment of Cell Mixture Distribution
3.7. Statistical Analysis, Dimension Reduction and Region-Level Methylation Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [Green Version]
- Chung, F.F.L.; Herceg, Z. The promises and challenges of toxico-epigenomics: Environmental chemicals and their impacts on the epigenome. Environ. Health Perspect. 2020, 128, e015001. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.-H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.-P.; Baylin, P.A.J.S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and can-cer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef]
- Ling, C. Epigenetic regulation of insulin action and secretion—Role in the pathogenesis of type 2 diabetes. J. Intern. Med. 2020, 288, 158–167. [Google Scholar] [CrossRef]
- Stanzione, R.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Forte, M.; Volpe, M.; Rubattu, S. Pathogenesis of ischemic stroke: Role of epi-genetic mechanisms. Genes 2020, 11, e89. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental Origins of Health and Disease: Brief History of the Approach and Current Focus on Epigenetic Mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Küpers, L.; Monnereau, C.; Sharp, G.; Yousefi, P.; Salas, L.; Ghantous, A.; Page, C.; Reese, R.; Willcox, A.; Czamara, D.; et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat. Commun. 2019, 10, 1893. [Google Scholar] [CrossRef] [Green Version]
- IARC. Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene; IARC: Lyon, France, 2002; Volume 82, pp. 1–556. [Google Scholar]
- Routledge, M.N.; Kimanya, M.E.; Shirima, C.P.; Wild, C.P.; Gong, Y.Y. Quantitative correlation of aflatoxin biomarker with dietary intake of aflatoxin in Tanzanian children. Biomarkers 2014, 19, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Shirima, C.P.; Kimanya, M.E.; Routledge, M.N.; Srey, C.; Kinabo, J.L.; Humpf, H.-U.; Wild, C.P.; Tu, Y.-K.; Gong, Y.Y. A Prospective Study of Growth and Biomarkers of Exposure to Aflatoxin and Fumonisin during Early Childhood in Tanzania. Environ. Health Perspect. 2015, 123, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.; Gong, Y.Y.; Routledge, M.N. Interventions Targeting Child Undernutrition in Developing Countries May Be Undermined by Dietary Exposure to Aflatoxin. Crit. Rev. Food Sci. Nutr. 2017, 57, 1963–1975. [Google Scholar] [CrossRef]
- Xu, Y.; Gong, Y.; Routledge, M. Aflatoxin exposure assessed by aflatoxin albumin adduct biomarker in populations from six African countries. World Mycotoxin J. 2018, 11, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Kamala, A.; Shirima, C.; Jani, B.; Bakari, M.; Sillo, H.; Rusibamayila, N.; De Saeger, S.; Kimanya, M.; Gong, Y.; Simba, A.; et al. Outbreak of an acute aflatoxicosis in Tanzania during 2016. World Mycotoxin J. 2018, 11, 311–320. [Google Scholar] [CrossRef]
- Gong, Y.Y.; Watson, S.; Routledge, M.N. Aflatoxin Exposure and Associated Human Health Effects, a Review of Epidemiological Studies. Food Saf. 2016, 4, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.; Moore, S.E.; Darboe, M.K.; Chen, G.; Tu, Y.-K.; Huang, Y.-T.; Eriksen, K.G.; Bernstein, R.M.; Prentice, A.M.; Wild, C.P.; et al. Impaired growth in rural Gambian infants exposed to aflatoxin: A prospective cohort study. BMC Public Health 2018, 18, 1247. [Google Scholar] [CrossRef]
- Aguilar, F.; Hussain, S.P.; Cerutti, P. Aflatoxin B1 induces the transversion of G-T in codon 249 of the P53 tumour suppressor gene in human hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 8586–8590. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Vargas, H.; Castelino, J.; Silver, M.J.; Dominguez-Salas, P.; Cros, M.-P.; Durand, G.; Le Calvez-Kelm, F.; Prentice, A.M.; Wild, C.P.; Moore, S.E.; et al. Exposure to aflatoxin B1 in utero is associated with DNA meth-ylation in white blood cells of infants in The Gambia. Int. J. Epidemiol. 2015, 44, 1238–1248. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.E.; Fulford, A.J.; Darboe, M.K.; Jobarteh, M.L.; Jarjou, L.M.; Prentice, A.M. A randomized trial to investigate the effects of pre-natal and infant nutritional supplementation on infant immune development in rural Gambia: The ENID trial: Early Nutrition and Immune Development. BMC Pregnancy Childbirth 2012, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Chapot, B.; Wild, C.P. ELISA for quantification of aflatoxin-albumin adducts and their application to human exposure as-sessment. Tech. Diagn. Pathol. 1991, 2, 135–155. [Google Scholar]
- Winkler, J.; Xu, Y.; Gong, Y.Y.; Lindahl, J.; Kersten, S.; Dänicke, S.; Routledge, M.N. Preliminary study on the relationship between af-latoxin-bovine serum albumin adducts in blood and aflatoxin M1 levels in milk of dairy cows. Mycotoxin Res. 2019, 36, 207–211. [Google Scholar]
- McCoy, L.F.; Scholl, P.F.; Sutcliffe, A.E.; Kieszak, S.M.; Powers, C.D.; Rogers, H.S.; Gong, Y.Y.; Groopman, J.D.; Wild, C.P.; Schleicher, R.L. Hu-man aflatoxin albumin adducts quantitatively compared by ELISA, HPLC with fluourescence detection, and HPLC with isotope dilution mass spectrometry. Cancer Epidemiol. Prev. Biomark. 2008, 17, 1653–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNAmethylation rray with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehen-sive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.S.; Bravo, H.C.; Leek, J.T. Removing batch effects for prediction problems with frozen surrogate variable analysis. PeerJ 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Perrier, F.; Novaloaca, A.; Ambatipudi, S.; Baglietto, L.; Ghantous, A.; Perduca, V.; Barrdahl, M.; Harlid, S.; Ong, K.K.; Cardona, A.; et al. Identi-Fying and correcting epigenetics measurements for systematic sources of variation. Clin. Epigenetics 2018, 10, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.C.; Barton, S.; Holbrook, J.D. How to make DNA methylome wide association studies more powerful. Epigenomics 2016, 8, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Reinius, L.E.; Acevedo, N.; Joerink, M.; Pershagen, G.; Dahlén, S.E.; Greco, D.; Söderhäll, C.; Scheynius, A.; Kere, J. Differential DNA methylation in purified human blood cells: Implications for cell lineage and studies on disease susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar] [CrossRef]
- Kaushal, A.; Zhang, H.; Karmaus, W.J.J.; Ray, M.; Torres, M.A.; Smith, A.K.; Wang, S.-L. Comparison of different cell type correction methods for genome-scale epigenetics studies. BMC Bioinform. 2017, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.A.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.F.; Lin, S.M. Comparison of Beta-value and M-value methods for quan-tifying methylation levels by microarray analysis. BMC Bioinormatics 2010, 11, 587. [Google Scholar]
- Ambatipudi, S.; Cuenin, C.; Hernandez-Vargas, H.; Ghantous, A.; Le Calvez-Kelm, F.; Kaaks, R.; Barrdahl, M.; Boeing, H.; Aleksan-drova, K.; Trichopoulou, A.; et al. Tobacco smoking-associated ge-nome-wide DNA methylation changes in the EPIC study. Epigenomics 2016, 8, 599–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.D.; Fernandez-Jimenez, N.; Ghantous, A.; degli Esposti, D.; Cuenin, C.; Cahais, V.; Choi, I.J.; Kim, Y.-I.; Kim, J.; Herceg, Z. Genome-wide profiling of normal gastric mucosa identifiesHelicobacter pylori- and cancer-associated DNA methylome changes. Int. J. Cancer 2018, 143, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wild, C.P.; Hudson, G.J.; Sabbioni, G.; Chapot, B.; Hall, A.J.; Wogan, G.N.; Whittle, H.; Montesano, R.; Groopman, J.D. Dietary intake of aflatoxins and the level of albumin-bound aflatoxin in peripheral blood in The Gambia, West Africa. Cancer Epidemiol. Prev. Biomark. 1992, 1, 229–234. [Google Scholar]
- Castelino, J.M.; Dominguez-Salas, P.; Routledge, M.N.; Prentice, A.M.; Moore, S.E.; Hennig, B.J.; Wild, C.P.; Gong, Y.Y. Seasonal and gesta-tion stage associated differences in aflatoxin exposure in pregnant Gambian women. Trop. Med. Int. Health 2014, 19, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.Y.; Hounsa, A.; Egal, S.; Turner, P.C.; Sutcliffe, A.E.; Hall, A.J.; Cardwell, K.; Wild, C.P. Postweaning Exposure to Aflatoxin Results in Impaired Child Growth: A Longitudinal Study in Benin, West Africa. Environ. Health Perspect. 2004, 112, 1334–1338. [Google Scholar] [CrossRef]
- Gong, Y.Y.; Wilson, S.; Mwatha, J.K.; Routledge, M.N.; Castelino, J.M.; Zhao, B.; Kimani, G.; Kariuki, H.C.; Vennervald, B.; Dunne, D.; et al. Aflatoxin Exposure May Contribute to Chronic Hepatomegaly in Kenyan School Children. Environ. Health Perspect. 2012, 120, 893–896. [Google Scholar] [CrossRef]
- Castelino, J.M.; Routledge, M.N.; Wilson, S.; Dunne, D.W.; Mwatha, J.K.; Gachuhi, K.; Wild, C.P.; Gong, Y.Y. Aflatoxin exposure is inversely associated with IGF1 and IGFBP3 levels in vitro and in Kenyan schoolchildren. Mol. Nutr. Food Res. 2015, 59, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; et al. Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef]
- Turner, P.C.; Moore, S.E.; Hall, A.J.; Prentice, A.M.; Wild, C.P. Modification of immune function through exposure to dietary aflatoxin in Gambian children. Environ. Health Perspect. 2003, 111, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.E.; Prendergast, A.J.; Turner, P.C.; Humphrey, J.H.; Stoltzfus, R.J. Aflatoxin exposure during pregnancy, maternal anemia, and adverse birth outcomes. Am. J. Trop. Med. Hyg. 2017, 96, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksen, K.G.; Johnson, W.; Sonko, B.; Prentice, A.M.; Darboe, M.K.; Moore, S.E. Following the World health Organization’s recom-mendation of exclusive breastfeeding to 6 months of age does not impact the growth of rural Gambian infants. J. Nutr. 2017, 147, 248–255. [Google Scholar] [CrossRef]
- Zarba, A.; Wild, C.P.; Hudson, G.J.; Hall, A.J.; Montesano, R.; Groopman, J.D. Aflatoxin M1 in human breast milk from The Gambia, West Africa, quantified by combined monoclonal antibody immunoaffinity chromatography and HPLC. Carcinogenesis 1992, 13, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Y.; Egal, S.; Hounsa, A.; Turner, P.C.; Hall, A.J.; Cardwell, K.F.; Wild, C.P. Determinants of aflatoxin exposure in young children from Benin and Togo, West Africa: The critical role of weaning. Int. J. Epidemiol. 2003, 32, 556–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.H.; Chiu, W.Z.; Breedveld, G.J.; Li, K.W.; Verkerk, A.J.M.H.; Hondius, D.; Hukema, R.; Seelaar, H.; Frick, P.; Severijnen, L.-A.; et al. PRKAR1B mutation associated with a new neurodegenerative disorder with unique pathology. Brain 2014, 137, 1361–1373. [Google Scholar] [CrossRef]
- Rieswijk, L.; Claessen, S.M.H.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S.; van Breda, S.G.J. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular car-cinoma. Toxicology 2016, 350, 31–39. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Li, D.; Zhang, B.; Li, M.; Li, W.; Zhu, W.; Xing, X.; Zeng, X.; Dong, G.; et al. Aberrant methylation of RUNX3 is present in aflatoxin B1-induced transformation of the LO2R cell line. Toxicology 2017, 385, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Soni, P.; Ghufran, M.S.; Kanade, S.R. Aflatoxin B1 induced multiple epigenetic modulators in human epithelial cell lines. Toxicon 2018, 151, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Trydyak, V.; Borowa-Mazgay, B.; Beland, F.A.; Pogribny, I.P. Gene expression and cytosine DNA methylation alterations in in-duced pluripotent stem cell-derived human hepatocytes treated with low doses of chemical carcinogens. Arch. Toxicol. 2019, 93, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gan, F.; Hou, L.; Liu, Z.; Su, J.; Lin, Z.; Le, G.; Huang, K. Aflatoxin B1 induces immunotoxicity through the DNA methyltrans-ferase-mediated JAK2/STAT3 pathway in 3D4/21 cells. J. Agric. Food Chem. 2019, 67, 3772–3780. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Naeem, I.; Gong, Y.Y.; Routledge, M.N.; Akhtar, S.; Riaz, M.; Ramalho, L.N.Z.; de Oliveira, C.A.F.; Ismail, Z. Early life exposure to dietary aflatoxins, health impact and control perspectives: A review. Trends Food Sci. Technol. 2021, 112, 212–224. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | n (%) |
|---|---|
| Total | 243 (100) |
| Male | 127 (52) |
| Female | 116 (48) |
| Ethnicity | |
| Fula | 16 (7) |
| Jola | 5 (2) |
| Mandinka | 222 (91) |
| Birth weight (kg) | Mean ± SD |
| 3.03 ± 0.38 | |
| AF-alb (pg/mg) | GM (95% CI) |
| 6 months (n = 230) | 3.78 (3.29, 4.34) |
| 12 months (n = 220) | 25.1 (21.67, 29.13) |
| 18 months (n = 210) | 49.48 (43.34, 56.49) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghantous, A.; Novoloaca, A.; Bouaoun, L.; Cuenin, C.; Cros, M.-P.; Xu, Y.; Hernandez-Vargas, H.; Darboe, M.K.; Prentice, A.M.; Moore, S.E.; et al. Aflatoxin Exposure during Early Life Is Associated with Differential DNA Methylation in Two-Year-Old Gambian Children. Int. J. Mol. Sci. 2021, 22, 8967. https://doi.org/10.3390/ijms22168967
Ghantous A, Novoloaca A, Bouaoun L, Cuenin C, Cros M-P, Xu Y, Hernandez-Vargas H, Darboe MK, Prentice AM, Moore SE, et al. Aflatoxin Exposure during Early Life Is Associated with Differential DNA Methylation in Two-Year-Old Gambian Children. International Journal of Molecular Sciences. 2021; 22(16):8967. https://doi.org/10.3390/ijms22168967
Chicago/Turabian StyleGhantous, Akram, Alexei Novoloaca, Liacine Bouaoun, Cyrille Cuenin, Marie-Pierre Cros, Ya Xu, Hector Hernandez-Vargas, Momodou K. Darboe, Andrew M. Prentice, Sophie E. Moore, and et al. 2021. "Aflatoxin Exposure during Early Life Is Associated with Differential DNA Methylation in Two-Year-Old Gambian Children" International Journal of Molecular Sciences 22, no. 16: 8967. https://doi.org/10.3390/ijms22168967
APA StyleGhantous, A., Novoloaca, A., Bouaoun, L., Cuenin, C., Cros, M. -P., Xu, Y., Hernandez-Vargas, H., Darboe, M. K., Prentice, A. M., Moore, S. E., Gong, Y. Y., Herceg, Z., & Routledge, M. N. (2021). Aflatoxin Exposure during Early Life Is Associated with Differential DNA Methylation in Two-Year-Old Gambian Children. International Journal of Molecular Sciences, 22(16), 8967. https://doi.org/10.3390/ijms22168967