
The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer

 , and
, and 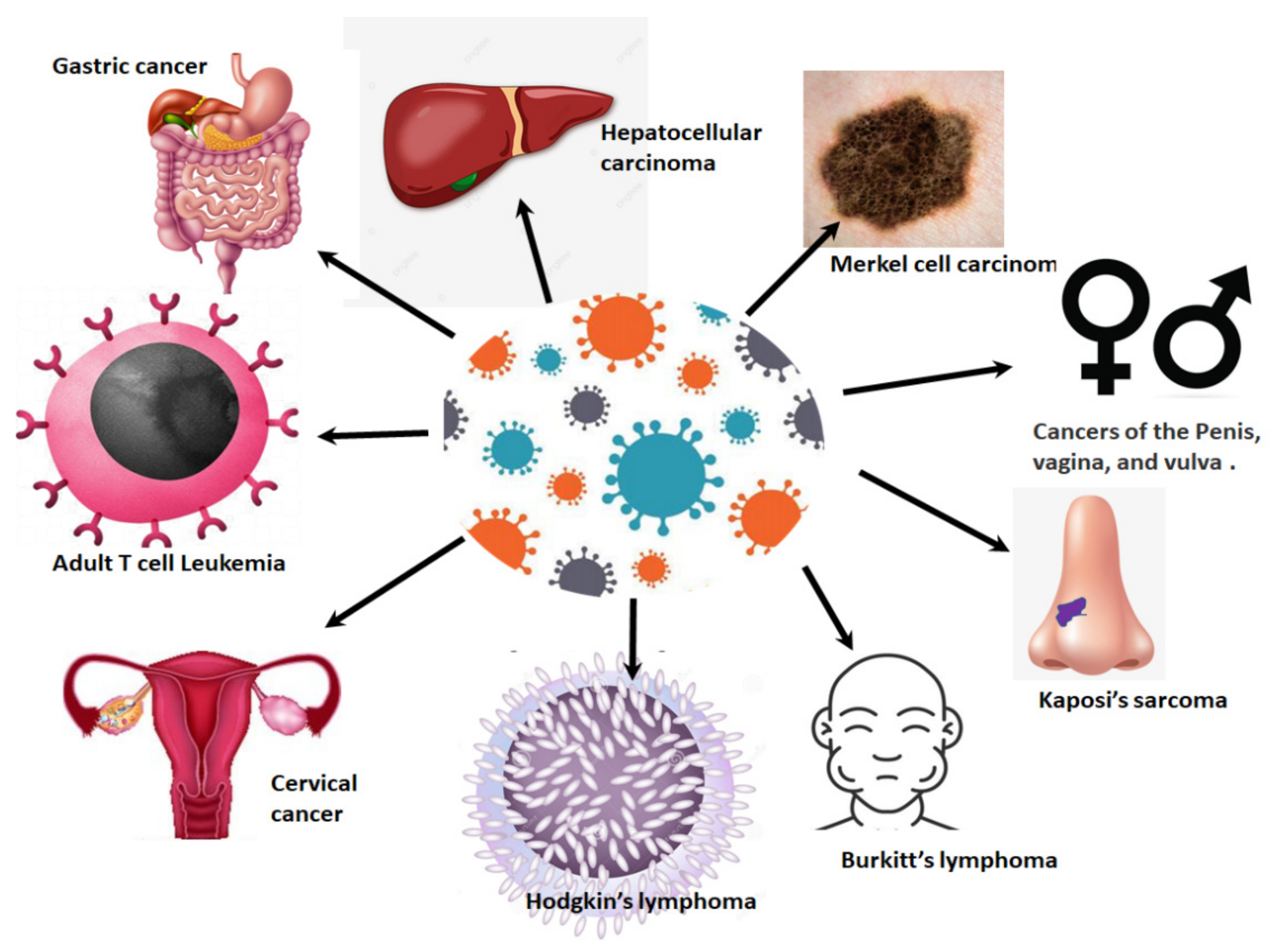{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
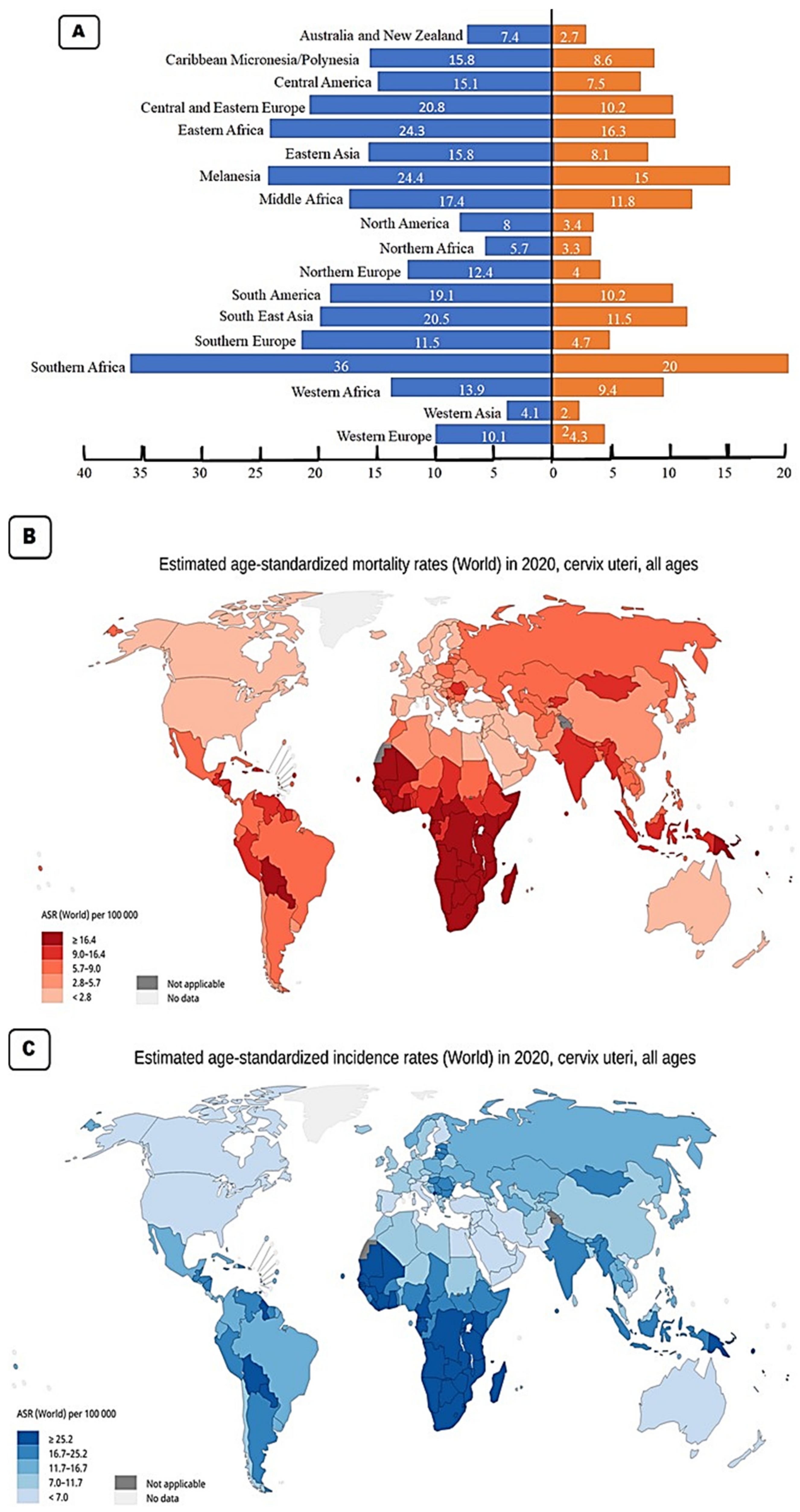
2. Cervical Cancer Epidemiology
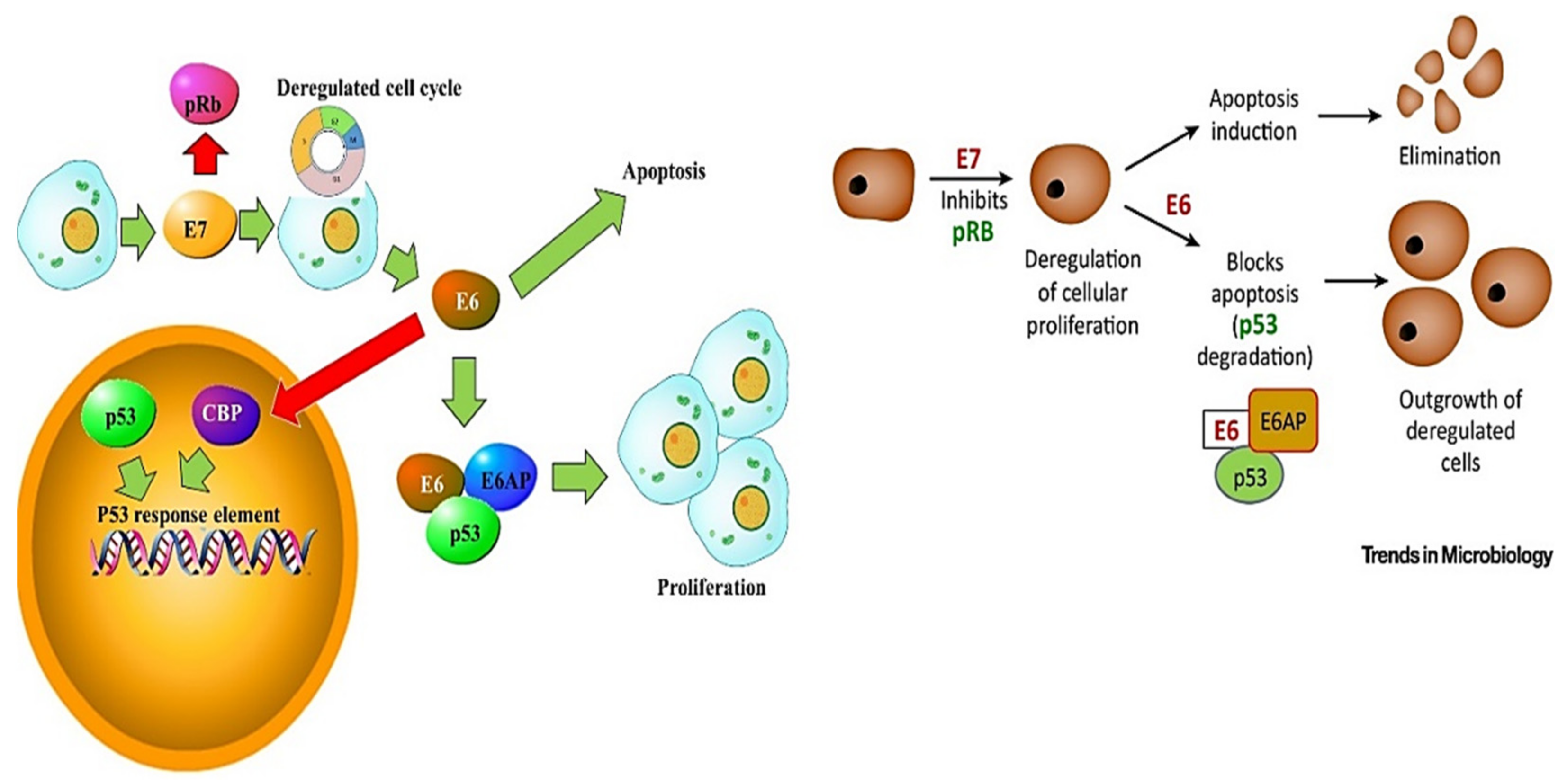
3. HPV Pathogenesis
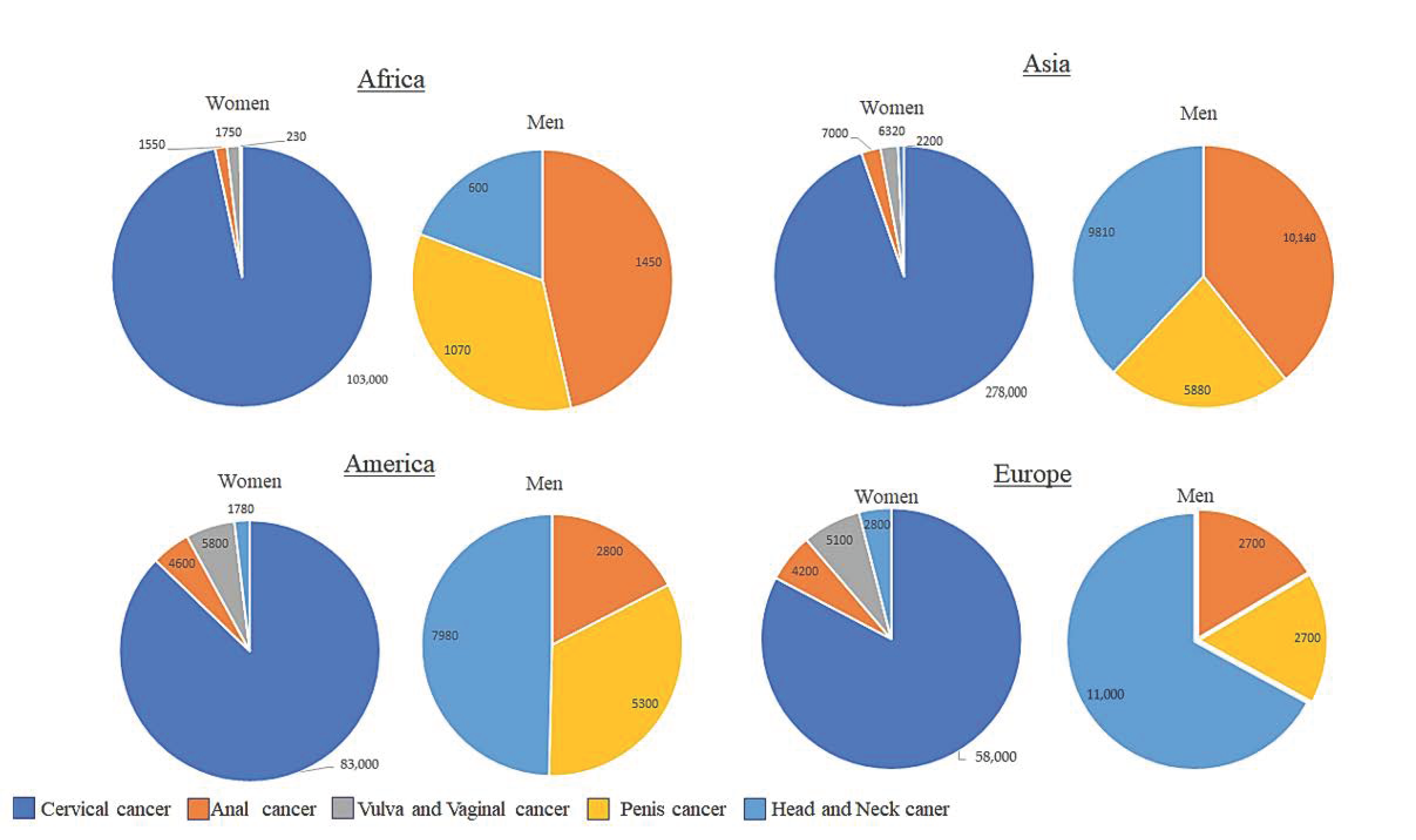
4. HPV-Related Cancers
5. Alternative Splicing
5.1. Alternative Splicing and Its Implications in Cervical Cancer
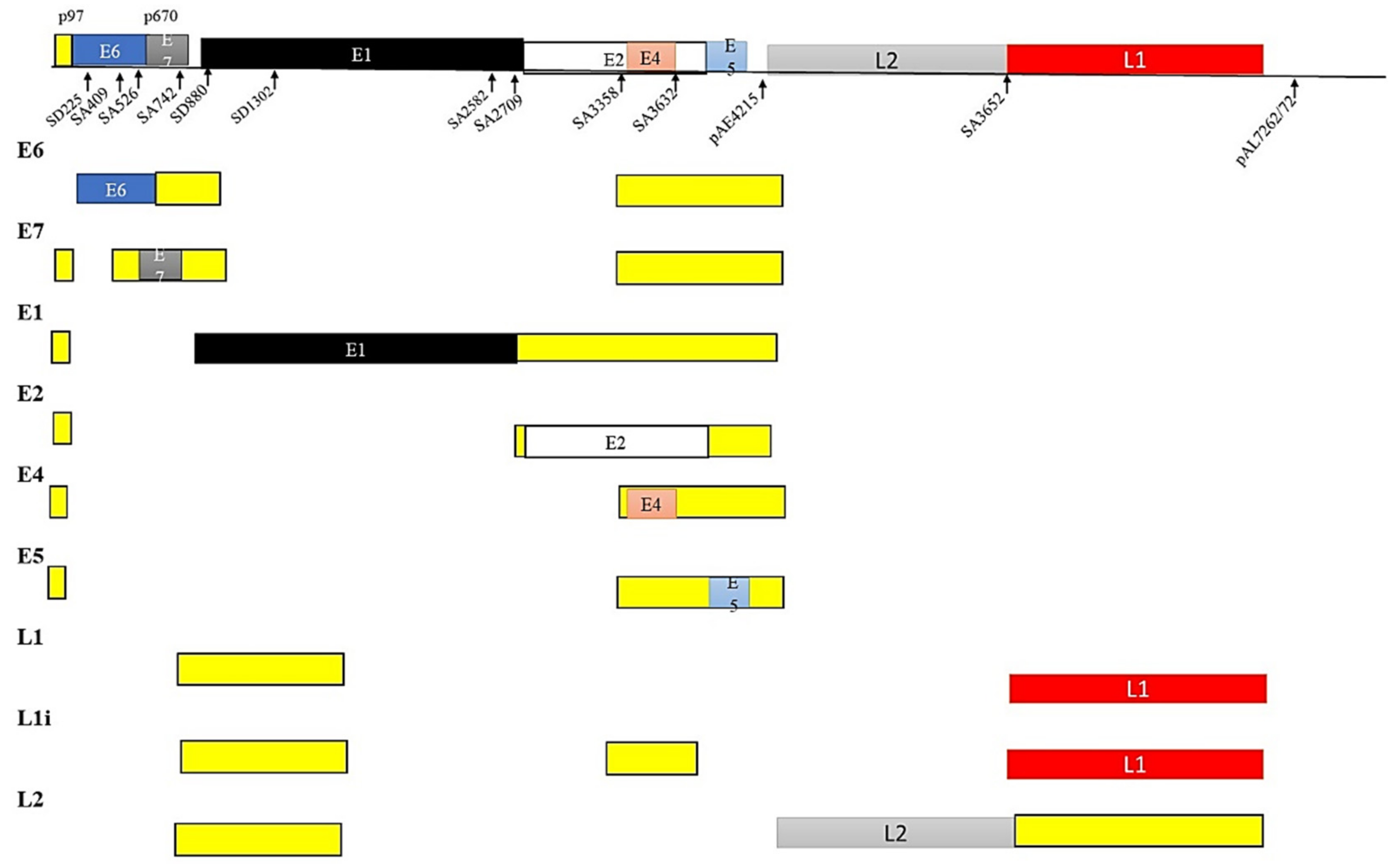
5.2. HPV and Splicing
5.3. HPV and Alternate mRNA Splicing
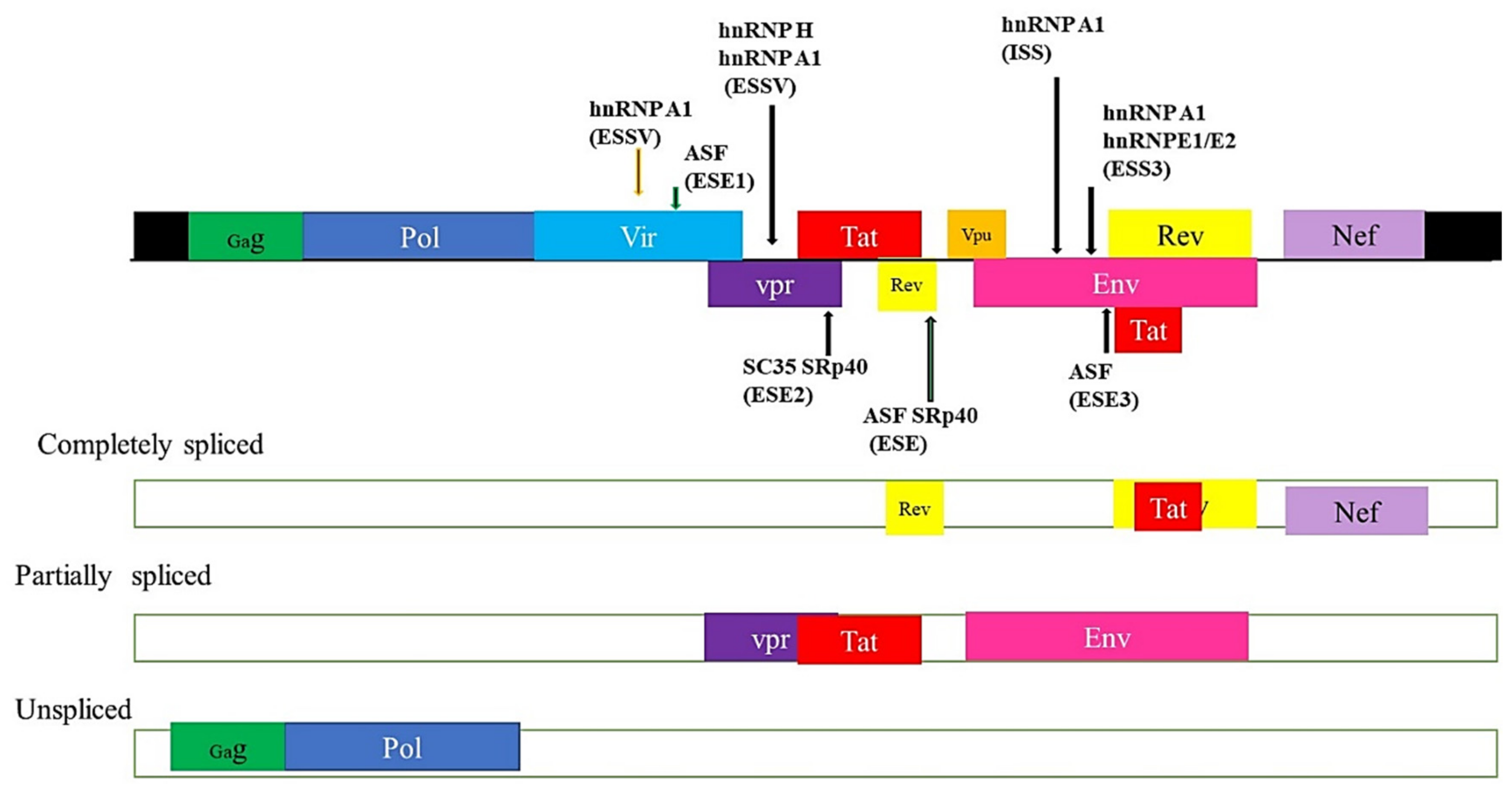
5.4. HIV and Alternate mRNA Splicing
5.5. RNA Splicing Factors
5.6. Splicing Factors Deregulation in Cervical Cancer
6. HPV, Genome Instability and DNA Damage Response (DDR)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hull, R.; Mbele, M.; Makhafola, T.; Hicks, C.; Wang, S.M.; Reis, R.M.; Mehrotra, R.; Mkhize-Kwitshana, Z.; Kibiki, G.; Bates, D.O.; et al. Cervical cancer in low and middle-income countries. Oncol. Lett. 2020, 20, 2058–2074. [Google Scholar] [CrossRef]
- Herrero, R.; González, P.; Markowitz, L.E. Present status of human papillomavirus vaccine development and implementation. Lancet Oncol. 2015, 16, e206–e216. [Google Scholar] [CrossRef]
- Francies, F.; Bassa, S.; Chatziioannou, A.; Kaufmann, A.; Dlamini, Z. Splicing Genomics Events in Cervical Cancer: Insights for Phenotypic Stratification and Biomarker Potency. Genes 2021, 12, 130. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Salehiniya, H. Cervical cancer in Iran: Integrative insights of epidemiological analysis. BioMedicine 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabekkodu, S.P.; Chakrabarty, S.; Ghosh, S.; Brand, A.; Satyamoorthy, K. Epigenomics, Pharmacoepigenomics, and Personalized Medicine in Cervical Cancer. Public Health Genom. 2017, 20, 100–115. [Google Scholar] [CrossRef]
- Akinlotan, M.; Bolin, J.N.; Helduser, J.; Ojinnaka, C.; Lichorad, A.; McClellan, D. Cervical Cancer Screening Barriers and Risk Factor Knowledge among Uninsured Women. J. Community Health 2017, 42, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.K.; Aimagambetova, G.; Ukybassova, T.; Kongrtay, K.; Azizan, A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination—Review of Current Perspectives. J. Oncol. 2019, 2019, 3257939. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R. HPV vaccination: The most pragmatic cervical cancer primary prevention strategy. Int. J. Gynaecol. Obstet. 2015, 131, S33–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joura, E.A.; Giuliano, A.R.; Iversen, O.E.; Bouchard, C.; Mao, C.; Mehlsen, J.; Moreira, E.D., Jr.; Ngan, Y.; Petersen, L.K.; Lazcano-Ponce, E.; et al. A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. N. Engl. J. Med. 2015, 372, 711–723. [Google Scholar] [CrossRef]
- Liu, F.; Dai, M.; Xu, Q.; Zhu, X.; Zhou, Y.; Jiang, S.; Wang, Y.; Ai, Z.; Ma, L.; Zhang, Y.; et al. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-κB-CD47 axis. Oncogene 2018, 37, 2394–2409. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Sharma, M.; Tan, N.; Barnabas, R.V. HIV-positive women have higher risk of human papilloma virus infection, precancerous lesions, and cervical cancer. AIDS 2018, 32, 795–808. [Google Scholar] [CrossRef]
- Bruni, L.; Albero, G.; Serrano, B.; Mena, M.; Gómez, D.; Muñoz, J. Human Papillomavirus and Related Diseases in the World—Summary Report. ICO/IARC Information Center in HPV and Cancer (HPV Information Centre). 2019. Available online: https://hpvcentre.net/statistics/reports/XWX.pdf (accessed on 30 June 2021).
- De Vuyst, H.; Alemany, L.; Lacey, C.; Chibwesha, C.J.; Sahasrabuddhe, V.; Banura, C.; Denny, L.; Parham, G.P. The Burden of Human Papillomavirus Infections and Related Diseases in Sub-Saharan Africa. Vaccine 2013, 31, F32–F46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, G.M.; Franceschi, S.; Keiser, O.; Schöni-Affolter, F.; Lise, M.; Dehler, S.; Levi, F.; Mousavi, M.; Bouchardy, C.; Wolfensberger, A.; et al. Immunodeficiency and the risk of cervical intraepithelial neoplasia 2/3 and cervical cancer: A nested case-control study in the Swiss HIV cohort study. Int. J. Cancer 2016, 138, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Strickler, H.D.; Burk, R.D.; Fazzari, M.; Anastos, K.; Minkoff, H.; Massad, L.S.; Hall, C.; Bacon, M.; Levine, A.M.; Watts, D.H.; et al. Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J. Natl. Cancer Inst. 2005, 97, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghill, A.E.; Shiels, M.S.; Suneja, G.; Engels, E.A. Elevated Cancer-Specific Mortality among HIV-Infected Patients in the United States. J. Clin. Oncol. 2015, 33, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Dryden-Peterson, S.; Bvochora-Nsingo, M.; Suneja, G.; Efstathiou, J.A.; Grover, S.; Chiyapo, S.; Ramogola-Masire, D.; Kebabonye-Pusoentsi, M.; Clayman, R.; Mapes, A.C.; et al. HIV Infection and Survival among Women with Cervical Cancer. J. Clin. Oncol. 2016, 34, 3749–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UNAIDS. Global AIDS Update 2017. Available online: http://www.unaids.org/sites/default/files/media_asset/global-AIDS-update-2016_en.pdf (accessed on 30 June 2021).
- Adler, D.H.; Kakinami, L.; Modisenyane, T.; Tshabangu, N.; Mohapi, L.; De Bruyn, G.; Martinson, N.A.; Omar, T. Increased regression and decreased incidence of human papillomavirus-related cervical lesions among HIV-infected women on HAART. AIDS 2012, 26, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Schiller, J.T.; Lowy, D.R. Virus Infection and Human Cancer: An Overview. Methods Mol. Biol. 2014, 193, 1–10. [Google Scholar]
- Moukassa, D.; Boumba, A.M.; Ngatali, C.F.; Ebatetou, A.; Mbon, J.B.N.; Ibara, J.-R. Virus-Induced Cancers in Africa: Epidemiology and Carcinogenesis Mechanisms. Open J. Pathol. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Habbema, D.; Weinmann, S.; Arbyn, M.; Kamineni, A.; Williams, A.E.; de Kok, I.M.C.M.; van Kemenade, F.; Field, T.S.; van Rosmalen, J.; Brown, M.L. Harms of cervical cancer screening in the United States and the Netherlands. Int. J. Cancer 2017, 140, 1215–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GLOBOCAN. Global Cervical Cancer Fact She. Available online: https://gco.iarc.fr/to¬day/data/factsheets/cancers/27-Cervix-uteri-fact-sheet.pdf (accessed on 27 May 2021).
- Bansal, A.; Singh, M.P.; Rai, B. Human papillomavirus-associated cancers: A growing global problem. Int. J. Appl. Basic Med Res. 2016, 6, 84–89. [Google Scholar] [PubMed] [Green Version]
- Francies, F.; Dlamini, Z. Aberrant Splicing Events and Epigenetics in Viral Oncogenomics: Current Therapeutic Strategies. Cells 2021, 10, 239. [Google Scholar] [CrossRef]
- Kelly, H.A.; Sawadogo, B.; Chikandiwa, A.; Segondy, M.; Gilham, C.; Lompo, O.; Omar, T.; Didelot, M.N.; Nagot, N.; Meda, N.; et al. Epidemiology of high-risk human papillomavirus and cervical lesions in African women living with HIV/AIDS: Effect of anti-retroviral therapy. AIDS 2017, 31, 273–285. [Google Scholar] [CrossRef]
- Cobucci, R.N.; Lima, P.H.; de Souza, P.C.; Costa, V.V.; Cornetta Mda, C.; Fernandes, J.V.; Gonçalves, A.K. Assessing the impact of HAART on the incidence of defining and non-defining AIDS cancers among patients with HIV/AIDS: A systematic review. J. Infect. Public Health 2015, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Firnhaber, C.; Westreich, D.; Schulze, D.; Williams, S.; Siminya, M.; Michelow, P.; Levin, S.; Faesen, M.; Smith, J.S. Highly active antiretroviral therapy and cervical dysplasia in HIV-positive women in South Africa. J. Int. AIDS Soc. 2012, 15, 17382. [Google Scholar] [CrossRef]
- Omar, T.; Schwartz, S.; Hanrahan, C.; Modisenyane, T.; Tshabangu, N.; Golub, J.E.; McIntyre, J.A.; Gray, G.E.; Mohapi, L.; Martinson, N.A. Progression and regression of premalignant cervical lesions in HIV-infected women from Soweto: A prospective cohort. AIDS 2011, 25, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrade, A.C.V.; Luz, P.M.; Velasque, L.; Veloso, V.G.; Moreira, R.I.; Russomano, F.; Chicarino-Coelho, J.; Pires, E.; Levi, J.E.; Grinsztejn, B.; et al. Factors Associated with Colposcopy-Histopathology Confirmed Cervical Intraepithelial Neoplasia among HIV-Infected Women from Rio De Janeiro, Brazil. PLoS ONE 2011, 6, e18297. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Brischiliari, S.C.; Gimenes, F.; de Abreu, A.L.; Irie, M.M.T.; Souza, R.P.; Santana, R.G.; Gravena, A.A.F.; Carvalho, M.D.d.B.; Consolaro, M.E.; Pelloso, S.M. Risk factors for cervical HPV infection and genotypes distribution in HIV-infected South Brazilian women. Infect. Agents Cancer 2014, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-Y.; Fei, M.-D.; Jiang, Y.; Fei, Q.-Y.; Qian, H.; Xu, L.; Jin, Y.-N.; Jiang, C.-Q.; Li, H.-X.; Tiggelaar, S.M.; et al. The diversity of human papillomavirus infection among human immunodeficiency virus-infected women in Yunnan, China. Virol. J. 2014, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Papillomaviruses, H. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2011. [Google Scholar]
- Dong, M.; Dong, Z.; Zhu, X.; Zhang, Y.; Song, L. Long non-coding RNA MIR205HG regulates KRT17 and tumor processes in cervical cancer via interaction with SRSF1. Exp. Mol. Pathol. 2019, 111, 104322. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, B.R.; Ryu, J.S.; Lee, G.O.; Kim, H.R.; Choi, K.H.; Ryu, J.W.; Na, K.S.; Park, M.C.; So, H.S.; et al. HNRNPA1, a Splicing Regulator, Is an Effective Target Protein for Cervical Cancer Detection: Comparison With Conventional Tumor Markers. Int. J. Gynecol. Cancer 2017, 27, 326–331. [Google Scholar] [CrossRef]
- Mole, S.; Faizo, A.A.A.; Hernandez-Lopez, H.; Griffiths, M.; Stevenson, A.; Roberts, S.; Graham, S.V. Human papillomavirus type 16 infection activates the host serine arginine protein kinase 1 (SRPK1)—Splicing factor axis. J. Gen. Virol. 2020, 101, 523–532. [Google Scholar] [CrossRef]
- Li, W.; Qi, Y.; Cui, X.; Huo, Q.; Zhu, L.; Zhang, A.; Tan, M.; Hong, Q.; Yang, Y.; Zhang, H.; et al. Characteristic of HPV Integration in the Genome and Transcriptome of Cervical Cancer Tissues. BioMed Res. Int. 2018, 2018, 6242173. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Contreras-Paredes, A.; Lizano, M. The Role of E6 Spliced Isoforms (E6*) in Human Papillomavirus-Induced Carcinogenesis. Viruses 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajiro, M.; Zheng, Z.M. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infect. 2014, 3, e63. [Google Scholar] [CrossRef] [PubMed]
- Brant, A.C.; Majerciak, V.; Moreira, M.A.M.; Zheng, Z.M. HPV18 Utilizes Two Alternative Branch Sites for E6*I Splicing to Produce E7 Protein. Virol. Sin. 2019, 34, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Cerasuolo, A.; Annunziata, C.; Tortora, M.; Starita, N.; Stellato, G.; Greggi, S.; Maglione, M.G.; Ionna, F.; Losito, S.; Botti, G.; et al. Comparative analysis of HPV16 gene expression profiles in cervical and in oropharyngeal squamous cell carcinoma. Oncotarget 2017, 8, 34070–34081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerasuolo, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. The Role of RNA Splicing Factors in Cancer: Regulation of Viral and Human Gene Expression in Human Papillomavirus-Related Cervical Cancer. Front. Cell Dev. Biol. 2020, 8, 474. [Google Scholar] [CrossRef]
- McFarlane, M.; MacDonald, A.I.; Stevenson, A.; Graham, S.V. Human Papillomavirus 16 Oncoprotein Expression Is Controlled by the Cellular Splicing Factor SRSF2 (SC35). J. Virol. 2015, 89, 5276–5287. [Google Scholar] [CrossRef] [Green Version]
- Bodaghi, S.; Jia, R.; Zheng, Z.M. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology 2009, 386, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V.; Faizo, A.A.A. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, E.L.; Brimacombe, C.L.; Hartley, M.; Bell, I.; Graham, S.; Roberts, S. Human papillomavirus type 1 E1^E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J. Virol. 2014, 88, 12599–12611. [Google Scholar] [CrossRef] [Green Version]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Wang, B.-D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeyens, A.; Naessens, E.; Van Nuffel, A.; Weening, K.E.; Reilly, A.-M.; Claeys, E.; Trypsteen, W.; Vandekerckhove, L.; Eyckerman, S.; Gevaert, K.; et al. HIV-1 Vpr N-terminal tagging affects alternative splicing of the viral genome. Sci. Rep. 2016, 6, 34573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Han, S.; Zheng, Y.; Planelles, V.; Lee, Y. The landscape of alternative splicing in HIV-1 infected CD4 T-cells. BMC Med Genom. 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.; Zhou, S.; Pollom, E.; Swanstrom, R. Characterizing HIV-1 Splicing by Using Next-Generation Sequencing. J. Virol. 2017, 91, e02515-16. [Google Scholar] [CrossRef] [Green Version]
- Sertznig, H.; Hillebrand, F.; Erkelenz, S.; Schaal, H.; Widera, M. Behind the scenes of HIV-1 replication: Alternative splicing as the dependency factor on the quiet. Virology 2018, 516, 176–188. [Google Scholar] [CrossRef]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV/AIDS 2017, 9, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Ocwieja, K.E.; Sherrill-Mix, S.; Mukherjee, R.; Custers-Allen, R.; David, P.; Brown, M.; Wang, S.; Link, D.R.; Olson, J.; Travers, K.; et al. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012, 40, 10345–10355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, Y.; Fu, L. Aberrant expression and regulatory network of splicing factor-SRSF3 in tumors. J. Cancer 2020, 11, 3502–3511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lv, R.; Guo, W.; Li, X. microRNA-802 inhibits cell proliferation and induces apoptosis in human cervical cancer by targeting serine/arginine-rich splicing factor 9. J. Cell. Biochem. 2019, 120, 10370–10379. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Ran, L.; Liu, Y.; Zhong, S.H.; Zhou, P.P.; Liao, M.X.; Fang, W. Knockdown of hnRNP A2/B1 inhibits cell proliferation, invasion and cell cycle triggering apoptosis in cervical cancer via PI3K/AKT signaling pathway. Oncol. Rep. 2018, 39, 939–950. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zheng, T.; Yu, J.; Zhou, L.; Wang, L. LncRNA XIST accelerates cervical cancer progression via upregulating Fus through competitively binding with miR-200a. Biomed. Pharmacother. 2018, 105, 789–797. [Google Scholar] [CrossRef]
- Banerjee, N.S.; Wang, H.K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [Green Version]
- Prati, B.; Marangoni, B.; Boccardo, E. Human papillomavirus and genome instability: From productive infection to cancer. Clin. 2018, 73, e539s. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, J.W.; Pitot, H.C.; Lambert, P.F. Loss of Dependence on Continued Expression of the Human Papillomavirus 16 E7 Oncogene in Cervical Cancers and Precancerous Lesions Arising in Fanconi Anemia Pathway-Deficient Mice. mBio 2016, 7, e00628-16. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rösl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Dürst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E990–E998. [Google Scholar] [CrossRef] [Green Version]
- Lekoane, K.M.B.; Kuupiel, D.; Mashamba-Thompson, T.P.; Ginindza, T.G. The interplay of HIV and human papillomavirus-related cancers in sub-Saharan Africa: Scoping review. Syst. Rev. 2020, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, K.; Wu, C.; Schwartz, S. Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation. Int. J. Mol. Sci. 2018, 19, 1735. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhan, P.; Feng, S.; Ji, H.; Tian, W.; Wang, M.; Cheng, C.; Song, B. SRSF6 regulates alternative splicing of genes involved in DNA damage response and DNA repair in HeLa cells. Oncol. Rep. 2020, 44, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Serrano, B.; Brotons, M.; Bosch, F.X.; Bruni, L. Epidemiology and burden of HPV-related disease. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 14–26. [Google Scholar] [CrossRef]
- World Health Organization. Human papillomavirus vaccines: WHO position paper, October 2014. Wkly. Epidemiol. Rec. Relev. Épidémiologique Hebd. 2014, 89, 465–491. [Google Scholar]
- Ouyang, D.; Yang, P.; Cai, J.; Sun, S.; Wang, Z. Comprehensive analysis of prognostic alternative splicing signature in cervical cancer. Cancer Cell Int. 2020, 20, 221. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Rätsch, G. Comprehensive Analysis of Alternative Splicing across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marima, R.; Hull, R.; Lolas, G.; Syrigos, K.N.; Kgoebane-Maseko, M.; Kaufmann, A.M.; Dlamini, Z. The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer. Int. J. Mol. Sci. 2021, 22, 10115. https://doi.org/10.3390/ijms221810115
Marima R, Hull R, Lolas G, Syrigos KN, Kgoebane-Maseko M, Kaufmann AM, Dlamini Z. The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer. International Journal of Molecular Sciences. 2021; 22(18):10115. https://doi.org/10.3390/ijms221810115
Chicago/Turabian StyleMarima, Rahaba, Rodney Hull, Georgios Lolas, Konstantinos N. Syrigos, Minah Kgoebane-Maseko, Andreas Martin Kaufmann, and Zodwa Dlamini. 2021. "The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer" International Journal of Molecular Sciences 22, no. 18: 10115. https://doi.org/10.3390/ijms221810115
APA StyleMarima, R., Hull, R., Lolas, G., Syrigos, K. N., Kgoebane-Maseko, M., Kaufmann, A. M., & Dlamini, Z. (2021). The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer. International Journal of Molecular Sciences, 22(18), 10115. https://doi.org/10.3390/ijms221810115