
Non-Canonical Roles of Tau and Their Contribution to Synaptic Dysfunction

 ,
,  ,
,
Abstract
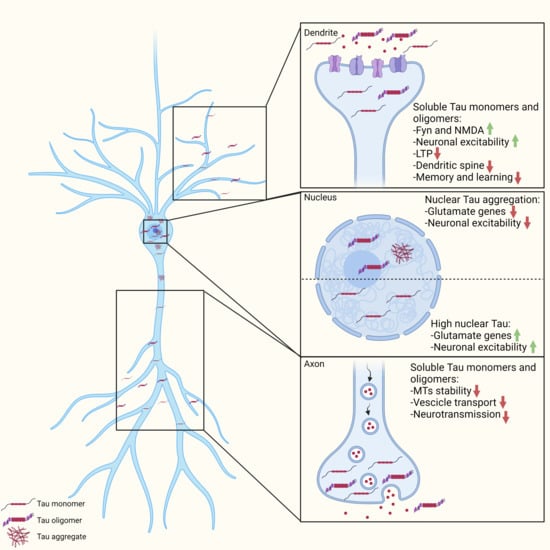
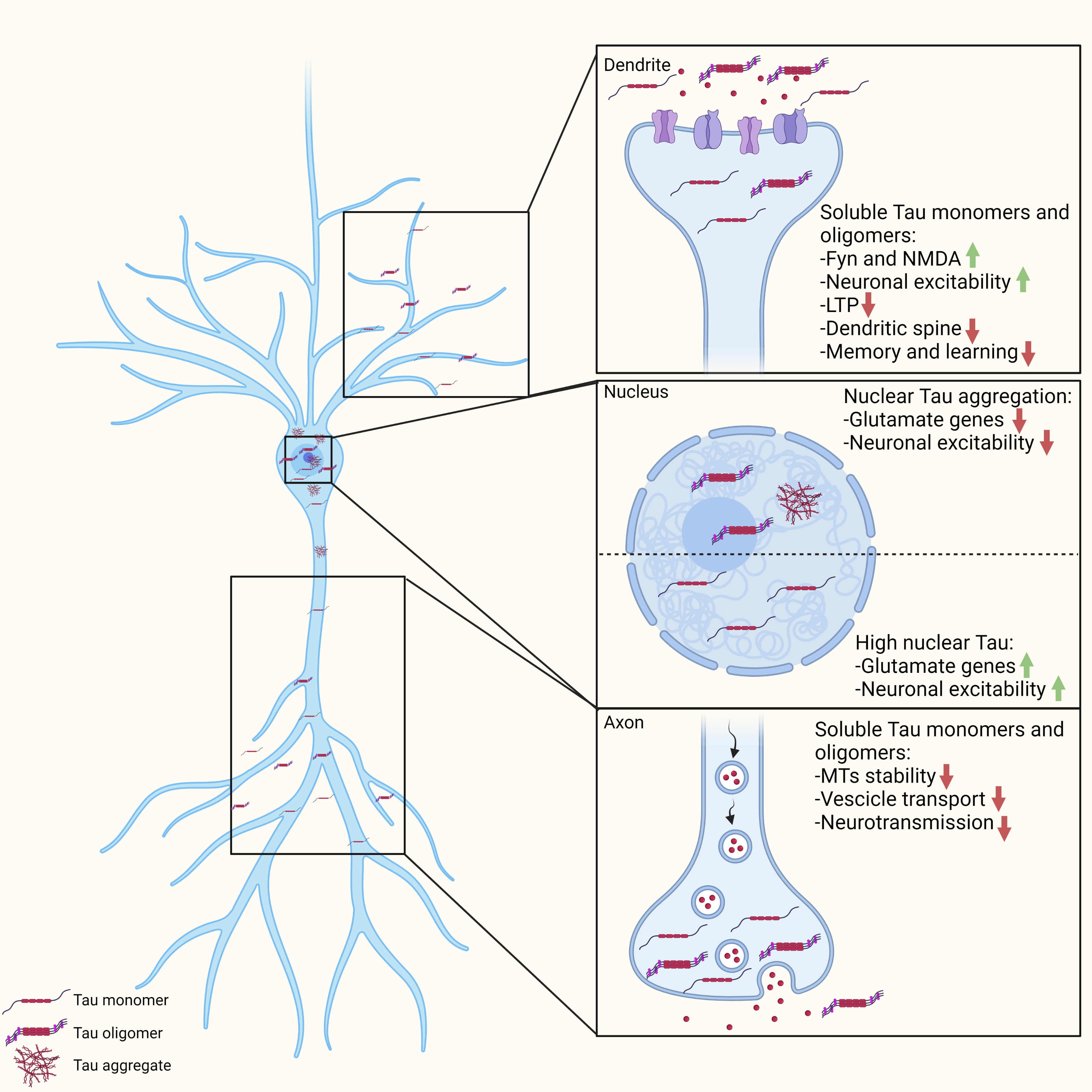
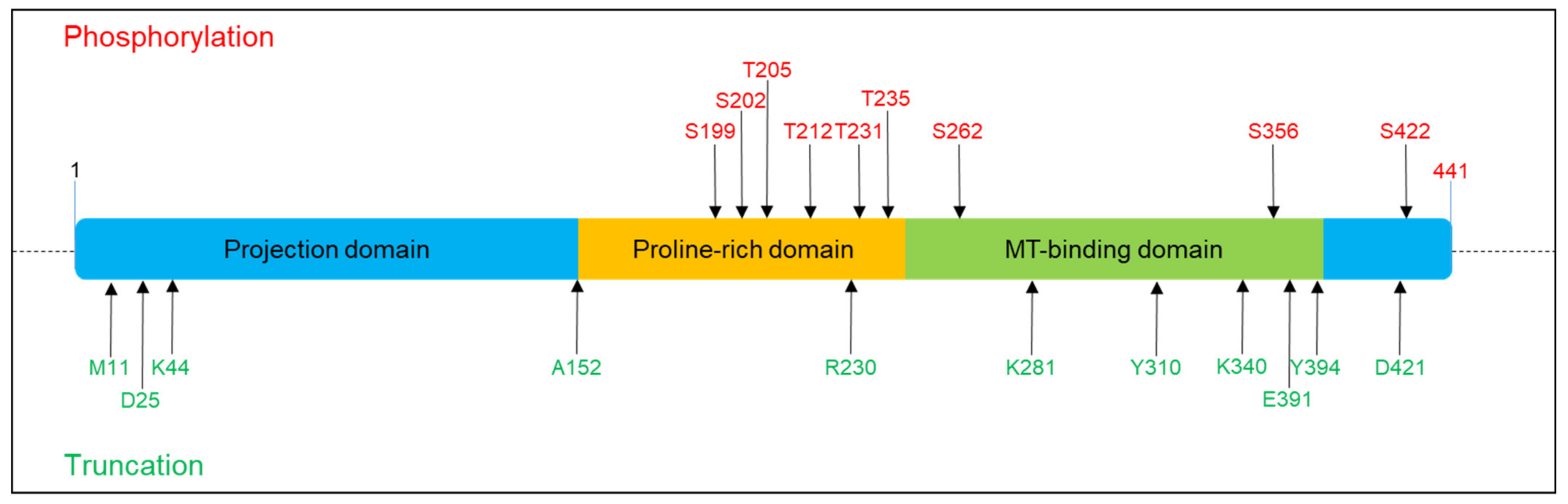
:
{kind=link}
{kind=link}
1. Tau Canonical Localization and Functions
2. Impact of Pathogenic Tau Localization
Tau Mislocalization in the Somato-Dendritic Compartment and Consequent Synaptic Dysfunction
3. Tau Functions in the Nuclear Compartment and Pathological Impact on Synaptic Functions
4. Tau-Mediated Gene Expression and Synaptic Dysfunction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Hirokawa, N.; Shiomura, Y.; Okabe, S. Tau proteins: The molecular structure and mode of binding on microtubules. J. Cell Biol. 1988, 107, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; Von Bergen, M.; Brutlach, H. Global Hairpin Folding of Tau in Solution †. Biochemistry 2006, 2283–2293. [Google Scholar] [CrossRef]
- Di Primio, C.; Quercioli, V.; Siano, G.; Rovere, M.; Kovacech, B.; Novak, M.; Cattaneo, A. The Distance between N and C Termini of Tau and of FTDP-17 Mutants Is Modulated by Microtubule Interactions in Living Cells. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.H.W.; Hogseth, M.; Phillips, E.C.; O’Neill, M.J.; Pooler, A.M.; Noble, W.; Hanger, D.P. Critical residues involved in tau binding to fyn: Implications for tau phosphorylation in Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Mondragón-Rodríguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kosik, K.S. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol. Cell 2001, 12, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [Green Version]
- Dotti, C.G.; Banker, G.A.; Binder, L.I. The expression and distribution of the microtubule-associated proteins tau and microtubule-associated protein 2 in hippocampal neurons in the rat in situ and in cell culture. Neuroscience 1987, 23, 121–130. [Google Scholar] [CrossRef]
- Mandell, J.W.; Banker, G.A. A spatial gradient of tau protein phosphorylation in nascent axons. J. Neurosci. 1996, 16, 5727–5740. [Google Scholar] [CrossRef]
- Pevalova, M.; Filipcik, P.; Novak, M.; Avila, J.; Iqbal, K. Post-translational modifications of tau protein. Bratisl. Lek. Listy 2006, 107, 346–353. [Google Scholar]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S.-I. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.D.; Di Clerico, J.; Li, B.; Corbo, C.P.; Alaniz, M.E.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J. Biol. Chem. 2010, 285, 30851–30860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraha, A.; Ghoshal, N.; Gamblin, T.C.; Cryns, V.; Berry, R.W.; Kuret, J.; Binder, L.I. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J. Cell Sci. 2000, 113 Pt 21, 3737–3745. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.d.C.; Mederlyova, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004, 279, 34873–34881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. 2005, 112, 813–838. [Google Scholar] [CrossRef]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef]
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Piedrahita, D.; Hernández, I.; López-Tobón, A.; Fedorov, D.; Obara, B.; Manjunath, B.S.; Boudreau, R.L.; Davidson, B.; Laferla, F.; Gallego-Gómez, J.C.; et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J. Neurosci. 2010, 30, 13966–13976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, G.; Roder, H.; Nunomura, A.; Takeda, A.; Friedlich, A.L.; Zhu, X.; Raina, A.K.; Holbrook, N.; Siedlak, S.L.; Harris, P.L.; et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10, 2411–2415. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Cho, J.-H.; Johnson, G.V.W. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem. 2003, 278, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Daly, N.L.; Hoffmann, R.; Otvos, L.; Craik, D.J. Role of phosphorylation in the conformation of tau peptides implicated in Alzheimer’s disease. Biochemistry 2000, 39, 9039–9046. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, S.; Klafki, H.W.; Plesnila, N.; Hübinger, G.; Obermeier, A.; Sahagún, H.; Monse, B.; Seneci, P.; Lewis, J.; Eriksen, J.; et al. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9673–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef]
- Fasulo, L.; Ugolini, G.; Visintin, M.; Bradbury, A.; Brancolini, C.; Verzillo, V.; Novak, M.; Cattaneo, A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000, 75, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Jakes, R.; Novak, M.; Davison, M.; Wischik, C.M. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J. 1991, 10, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [CrossRef]
- Novák, M. Truncated tau protein as a new marker for Alzheimer’s disease. Acta Virol. 1994, 38, 173–189. [Google Scholar]
- Cho, J.-H.; Johnson, G.V.W. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J. Biol. Chem. 2004, 279, 54716–54723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; LaPointe, N.; et al. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [Green Version]
- Ozcelik, S.; Sprenger, F.; Skachokova, Z.; Fraser, G.; Abramowski, D.; Clavaguera, F.; Probst, A.; Frank, S.; Müller, M.; Staufenbiel, M.; et al. Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol. Psychiatry 2016, 21, 1790–1798. [Google Scholar] [CrossRef] [Green Version]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Maria Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. J. Alzheimers. Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.-Y.; Trojanowski, J.Q. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, E.-J.; Liu, Q.; Li, S.-H.; Li, T.; Zhou, Q.-Z.; Liu, Y.-C.; Zhang, H.; Wang, J.-Z. Tau Acetylation in Entorhinal Cortex Induces its Chronic Hippocampal Propagation and Cognitive Deficits in Mice. J. Alzheimers. Dis. 2020, 77, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; David, J.P.; Lefranc, D.; Vermersch, P.; Wattez, A.; Delacourte, A. Different distribution of phosphorylated tau protein isoforms in Alzheimer’s and Pick’s diseases. FEBS Lett. 1997, 412, 578–582. [Google Scholar] [CrossRef] [Green Version]
- Delacourte, A.; Robitaille, Y.; Sergeant, N.; Buée, L.; Hof, P.R.; Wattez, A.; Laroche-Cholette, A.; Mathieu, J.; Chagnon, P.; Gauvreau, D. Specific pathological Tau protein variants characterize Pick’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 159–168. [Google Scholar] [CrossRef]
- Arai, T.; Ikeda, K.; Akiyama, H.; Shikamoto, Y.; Tsuchiya, K.; Yagishita, S.; Beach, T.; Rogers, J.; Schwab, C.; McGeer, P.L. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001, 101, 167–173. [Google Scholar] [CrossRef]
- Buée-Scherrer, V.; Hof, P.R.; Buée, L.; Leveugle, B.; Vermersch, P.; Perl, D.P.; Olanow, C.W.; Delacourte, A. Hyperphosphorylated tau proteins differentiate corticobasal degeneration and Pick’s disease. Acta Neuropathol. 1996, 91, 351–359. [Google Scholar] [CrossRef]
- Togo, T.; Sahara, N.; Yen, S.-H.; Cookson, N.; Ishizawa, T.; Hutton, M.; de Silva, R.; Lees, A.; Dickson, D.W. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Song, L.; Wells, E.A.; Robinson, A.S. Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines 2021, 9, 190. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.M.; Thomas, T.L.; Woodard, D.R.; Kashmer, O.M.; Diamond, M.I. Tau monomer encodes strains. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers. Dis. 2008, 14, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, J.; Deters, N.; Doldissen, A.; Bokhari, L.; Ke, Y.; Wiesner, A.; Schonrock, N.; Ittner, L.M. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007, 17, 91–103. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta - Mol. Basis Dis. 2005, 1739, 240–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Amos, L.A. Microtubule structure and its stabilisation. Org. Biomol. Chem. 2004, 2, 2153–2160. [Google Scholar] [CrossRef]
- Kar, S.; Fan, J.; Smith, M.J.; Goedert, M.; Amos, L.A. Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 2003, 22, 70–77. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers. Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Trinczek, B.; Ebneth, A.; Mandelkow, E.M.; Mandelkow, E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 1999, 112 Pt 1, 2355–2367. [Google Scholar] [CrossRef] [PubMed]
- Beevers, J.E.; Lai, M.C.; Collins, E.; Booth, H.D.E.; Zambon, F.; Parkkinen, L.; Vowles, J.; Cowley, S.A.; Wade-Martins, R.; Caffrey, T.M. MAPT Genetic Variation and Neuronal Maturity Alter Isoform Expression Affecting Axonal Transport in iPSC-Derived Dopamine Neurons. Stem Cell Reports 2017, 9, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-Z.; Rasenick, M.M. Tau associates with actin in differentiating PC12 cells. FASEB J. 2006, 20, 1452–1461. [Google Scholar] [CrossRef]
- Leugers, C.J.; Lee, G. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J. Biol. Chem. 2010, 285, 19125–19134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lante, F.; Buisson, A. Activity-Dependent Tau Protein Translocation to Excitatory Synapse Is Disrupted by Exposure to Amyloid-Beta Oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef]
- Tang, Z.; Ioja, E.; Bereczki, E.; Hultenby, K.; Li, C.; Guan, Z.; Winblad, B.; Pei, J.-J. mTor mediates tau localization and secretion: Implication for Alzheimer’s disease. Biochim. Biophys. Acta - Mol. Cell Res. 2015, 1853, 1646–1657. [Google Scholar] [CrossRef] [Green Version]
- Loomis, P.A.; Howard, T.H.; Castleberry, R.P.; Binder, L.I. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Futerman, A.H.; Banker, G.A. The economics of neurite outgrowth—The addition of new membrane to growing axons. Trends Neurosci. 1996, 19, 144–149. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: Focusing on phosphatases. FASEB J. 1995, 9, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.J.; Huber, B.R.; Silverman, M.A.; Cline, M.M.; Gill, T.B.; Cross, C.G.; Cook, D.G.; Minoshima, S. Intranasal Paclitaxel Alters Alzheimer’s Disease Phenotypic Features in 3xTg-AD Mice. J. Alzheimers. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Huang, Y.; Gao, Q.; Zhou, Q. Stabilization of microtubules improves cognitive functions and axonal transport of mitochondria in Alzheimer’s disease model mice. Neurobiol. Aging 2020, 96, 223–232. [Google Scholar] [CrossRef]
- Novak, P.; Kontsekova, E.; Zilka, N.; Novak, M. Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front. Neurosci. 2018, 12, 1–14. [Google Scholar] [CrossRef]
- Vitale, F.; Giliberto, L.; Ruiz, S.; Steslow, K.; Marambaud, P.; D’Abramo, C. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol. Commun. 2018, 6, 82. [Google Scholar] [CrossRef]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimers Dis. 2017, 4, 236–241. [Google Scholar]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense Reduction of Tau in Adult Mice Protects against Seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.-C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.J.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-Y.; Wei, Y.-P.; Xiong, Y.; Wang, X.-C.; Xie, A.-J.; Wang, X.-L.; Yang, Y.; Wang, Q.; Lu, Y.-M.; Liu, R.; et al. Synaptic Released Zinc Promotes Tau Hyperphosphorylation by Inhibition of Protein Phosphatase 2A (PP2A). J. Biol. Chem. 2012, 287, 11174–11182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Wei, Q.; Liu, F.-F.; Hu, F.; Xie, A.-J.; Zhu, L.-Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Pfister, S.; Notter, T.; Knuesel, I. Decisive role of Reelin signaling during early stages of Alzheimer’s disease. Neuroscience 2013, 246, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing dendritic spine pathology in Alzheimer’s disease: Problems and opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef] [Green Version]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers. Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Moreno, H.; Morfini, G.; Buitrago, L.; Ujlaki, G.; Choi, S.; Yu, E.; Moreira, J.E.; Avila, J.; Brady, S.T.; Pant, H.; et al. Tau pathology-mediated presynaptic dysfunction. Neuroscience 2016, 325, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.; Kitazawa, M.; Chabrier, M.A.; Cheng, D.; Baglietto-Vargas, D.; Kling, A.; Moeller, A.; Green, K.N.; LaFerla, F.M. Calpain Inhibitor A-705253 Mitigates Alzheimer’s Disease–Like Pathology and Cognitive Decline in Aged 3xTgAD Mice. Am. J. Pathol. 2012, 181, 616–625. [Google Scholar] [CrossRef]
- Roy, S.M.; Minasov, G.; Arancio, O.; Chico, L.W.; Van Eldik, L.J.; Anderson, W.F.; Pelletier, J.C.; Watterson, D.M. A Selective and Brain Penetrant p38αMAPK Inhibitor Candidate for Neurologic and Neuropsychiatric Disorders That Attenuates Neuroinflammation and Cognitive Dysfunction. J. Med. Chem. 2019, 62, 5298–5311. [Google Scholar] [CrossRef]
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.-W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90, 245–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickstein, D.L.; Brautigam, H.; Stockton, S.D.; Schmeidler, J.; Hof, P.R. Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct. Funct. 2010, 214, 161–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polydoro, M.; Acker, C.M.; Duff, K.; Castillo, P.E.; Davies, P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 2009, 29, 10741–10749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Bretteville, A.; Schindowski, K.; Gilissen, E.; Authelet, M.; De Decker, R.; Yilmaz, Z.; Buée, L.; Brion, J.-P. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am. J. Pathol. 2007, 171, 976–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef]
- Rady, R.M.; Zinkowski, R.P.; Binder, L.I. Presence of tau in isolated nuclei from human brain. Neurobiol. Aging 1995, 16, 479–486. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Lucas, J.J. Altered Levels and Isoforms of Tau and Nuclear Membrane Invaginations in Huntington’s Disease. Front. Cell. Neurosci. 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef] [Green Version]
- Rousseaux, M.W.C.; de Haro, M.; Lasagna-Reeves, C.A.; de Maio, A.; Park, J.; Jafar-Nejad, P.; Al-Ramahi, I.; Sharma, A.; See, L.; Lu, N.; et al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. Elife 2016, 5, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Diez, L.; Wegmann, S. Nuclear Transport Deficits in Tau-Related Neurodegenerative Diseases. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 1–21. [Google Scholar] [CrossRef]
- Dickson, J.R.; Yoon, H.; Frosch, M.P.; Hyman, B.T. Cytoplasmic Mislocalization of RNA Polymerase II Subunit RPB1 in Alzheimer Disease Is Linked to Pathologic Tau. J. Neuropathol. Exp. Neurol. 2021, 80, 530–540. [Google Scholar] [CrossRef]
- Mahoney, R.; Ochoa Thomas, E.; Ramirez, P.; Miller, H.E.; Beckmann, A.; Zuniga, G.; Dobrowolski, R.; Frost, B. Pathogenic Tau Causes a Toxic Depletion of Nuclear Calcium. Cell Rep. 2020, 32. [Google Scholar] [CrossRef]
- Chauderlier, A.; Gilles, M.; Spolcova, A.; Caillierez, R.; Chwastyniak, M.; Kress, M.; Drobecq, H.; Bonnefoy, E.; Pinet, F.; Weil, D.; et al. Tau/DDX6 interaction increases microRNA activity. Biochim. Biophys. Acta. Gene Regul. Mech. 2018, 1861, 762–772. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling Murine Tau with 0N, 1N and 2N Isoform-Specific Antibodies in Brain and Peripheral Organs Reveals Distinct Subcellular Localization, with the 1N Isoform Being Enriched in the Nucleus. PLoS One 2013, 8, e84849. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear Magnetic Resonance Spectroscopy Characterization of Interaction of Tau with DNA and Its Regulation by Phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef]
- Ulrich, G.; Salvadè, A.; Boersema, P.; Calì, T.; Foglieni, C.; Sola, M.; Picotti, P.; Papin, S.; Paganetti, P. Phosphorylation of nuclear Tau is modulated by distinct cellular pathways. Sci. Rep. 2018, 8, 17702. [Google Scholar] [CrossRef] [Green Version]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Q.; He, R.Q. Tau could protect DNA double helix structure. Biochim. Biophys. Acta - Proteins Proteomics 2003, 1645, 205–211. [Google Scholar] [CrossRef]
- Vasudevaraju, P.; Guerrero, E.; Hegde, M.L.; Collen, T.B.; Britton, G.B.; Rao, K.S. New evidence on α-synuclein and Tau binding to conformation and sequence specific GC* rich DNA: Relevance to neurological disorders. J. Pharm. Bioallied Sci. 2012, 4, 112–117. [Google Scholar] [PubMed]
- Sjöberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Benhelli-Mokrani, H.; Mansuroglu, Z.; Chauderlier, A.; Albaud, B.; Gentien, D.; Sommer, S.; Schirmer, C.; Laqueuvre, L.; Josse, T.; Buée, L.; et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. 2018, 1, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, Tau protein prevents DNA damage by peroxidation. PLoS One 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Redaelli, V.; Contiero, P.; Fabiano, S.; Tagliabue, G.; Perego, P.; Benussi, L.; Bruni, A.C.; Filippini, G.; Farinotti, M.; et al. Tau mutations serve as a novel risk factor for cancer. Cancer Res. 2018, 78, 3731–3739. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Conconi, D.; Panzeri, E.; Redaelli, S.; Piccoli, E.; Paoletta, L.; Dalprà, L.; Tagliavini, F. Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J. Alzheimer’s Dis. 2013, 33, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.; Conconi, D.; Panzeri, E.; Paoletta, L.; Piccoli, E.; Ferretti, M.G.; Mangieri, M.; Ruggerone, M.; Dalprà, L.; Tagliavini, F. Mutations in MAPT give rise to aneuploidy in animal models of tauopathy. Neurogenetics 2014, 15, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Gómez de Barreda, E.; Dawson, H.N.; Vitek, M.P.; Avila, J. Tau deficiency leads to the upregulation of BAF-57, a protein involved in neuron-specific gene repression. FEBS Lett. 2010, 584, 2265–2270. [Google Scholar] [CrossRef] [Green Version]
- Oyama, F.; Kotliarova, S.; Harada, A.; Ito, M.; Miyazaki, H.; Ueyama, Y.; Hirokawa, N.; Nukina, N.; Ihara, Y. Gem GTPase and tau: Morphological changes induced by gem GTPase in cho cells are antagonized by tau. J. Biol. Chem. 2004, 279, 27272–27277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Maina, M.B.; Bailey, L.J.; Doherty, A.J.; Serpell, L.C. The Involvement of Aβ42 and Tau in Nucleolar and Protein Synthesis Machinery Dysfunction. Front. Cell. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Portillo, M.; Eremenko, E.; Kaluski, S.; Garcia-Venzor, A.; Onn, L.; Stein, D.; Slobodnik, Z.; Zaretsky, A.; Ueberham, U.; Einav, M.; et al. SIRT6-CBP-dependent nuclear Tau accumulation and its role in protein synthesis. Cell Rep. 2021, 35, 109035. [Google Scholar] [CrossRef]
- Siano, G.; Varisco, M.; Caiazza, M.C.; Quercioli, V.; Mainardi, M.; Ippolito, C.; Cattaneo, A.; Di Primio, C. Tau Modulates VGluT1 Expression. J. Mol. Biol. 2019, 431, 873–884. [Google Scholar] [CrossRef]
- Siano, G.; Caiazza, M.C.; Varisco, M.; Calvello, M.; Quercioli, V.; Cattaneo, A.; Di Primio, C. Modulation of tau subcellular localization as a tool to investigate the expression of disease-related genes. J. Vis. Exp. 2019, 154, e59988. [Google Scholar] [CrossRef]
- Khadrawyb YA, E.H. Glutamate Excitotoxicity and Neurodegeneration. J. Mol. Genet. Med. 2014, 8, 8–11. [Google Scholar] [CrossRef]
- Crescenzi, R.; DeBrosse, C.; Nanga, R.P.R.; Byrne, M.D.; Krishnamoorthy, G.; D’Aquilla, K.; Nath, H.; Morales, K.H.; Iba, M.; Hariharan, H.; et al. Longitudinal imaging reveals sub-hippocampal dynamics in glutamate levels associated with histopathologic events in a mouse model of tauopathy and healthy mice. Hippocampus 2017, 27, 285–302. [Google Scholar] [CrossRef] [Green Version]
- Kazim, S.F.; Chuang, S.C.; Zhao, W.; Wong, R.K.S.; Bianchi, R.; Iqbal, K. Early-onset network hyperexcitability in presymptomatic Alzheimer’s disease transgenic mice is suppressed by passive immunization with anti-human APP/Aβ antibody and by mGluR5 blockade. Front. Aging Neurosci. 2017, 9, 1–17. [Google Scholar] [CrossRef]
- Angulo, S.L.; Orman, R.; Neymotin, S.A.; Liu, L.; Buitrago, L.; Cepeda-Prado, E.; Stefanov, D.; Lytton, W.W.; Stewart, M.; Small, S.A.; et al. Tau and amyloid-related pathologies in the entorhinal cortex have divergent effects in the hippocampal circuit. Neurobiol. Dis. 2017, 108, 261–276. [Google Scholar] [CrossRef]
- Sánchez, M.P.; García-Cabrero, A.M.; Sánchez-Elexpuru, G.; Burgos, D.F.; Serratosa, J.M. Tau-induced pathology in epilepsy and dementia: Notions from patients and animal models. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid β peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siano, G.; Varisco, M.; Scarlatti, A.; Caiazza, M.C.; Dunville, K.; Cremisi, F.; Costa, M.; Pancrazi, L.; Di Primio, C.; Cattaneo, A. Gene Expression of Disease-related Genes in Alzheimer’s Disease is Impaired by Tau Aggregation. J. Mol. Biol. 2020, 432, 166675. [Google Scholar] [CrossRef]
- Poirel, O.; Mella, S.; Videau, C.; Ramet, L.; Davoli, M.A.; Herzog, E.; Katsel, P.; Mechawar, N.; Haroutunian, V.; Epelbaum, J.; et al. Moderate decline in select synaptic markers in the prefrontal cortex (BA9) of patients with Alzheimer’s disease at various cognitive stages. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef]
- Kashani, A.; Lepicard, E.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J.; et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siano, G.; Falcicchia, C.; Origlia, N.; Cattaneo, A.; Di Primio, C. Non-Canonical Roles of Tau and Their Contribution to Synaptic Dysfunction. Int. J. Mol. Sci. 2021, 22, 10145. https://doi.org/10.3390/ijms221810145
Siano G, Falcicchia C, Origlia N, Cattaneo A, Di Primio C. Non-Canonical Roles of Tau and Their Contribution to Synaptic Dysfunction. International Journal of Molecular Sciences. 2021; 22(18):10145. https://doi.org/10.3390/ijms221810145
Chicago/Turabian StyleSiano, Giacomo, Chiara Falcicchia, Nicola Origlia, Antonino Cattaneo, and Cristina Di Primio. 2021. "Non-Canonical Roles of Tau and Their Contribution to Synaptic Dysfunction" International Journal of Molecular Sciences 22, no. 18: 10145. https://doi.org/10.3390/ijms221810145
APA StyleSiano, G., Falcicchia, C., Origlia, N., Cattaneo, A., & Di Primio, C. (2021). Non-Canonical Roles of Tau and Their Contribution to Synaptic Dysfunction. International Journal of Molecular Sciences, 22(18), 10145. https://doi.org/10.3390/ijms221810145