
Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition

 , and
, and 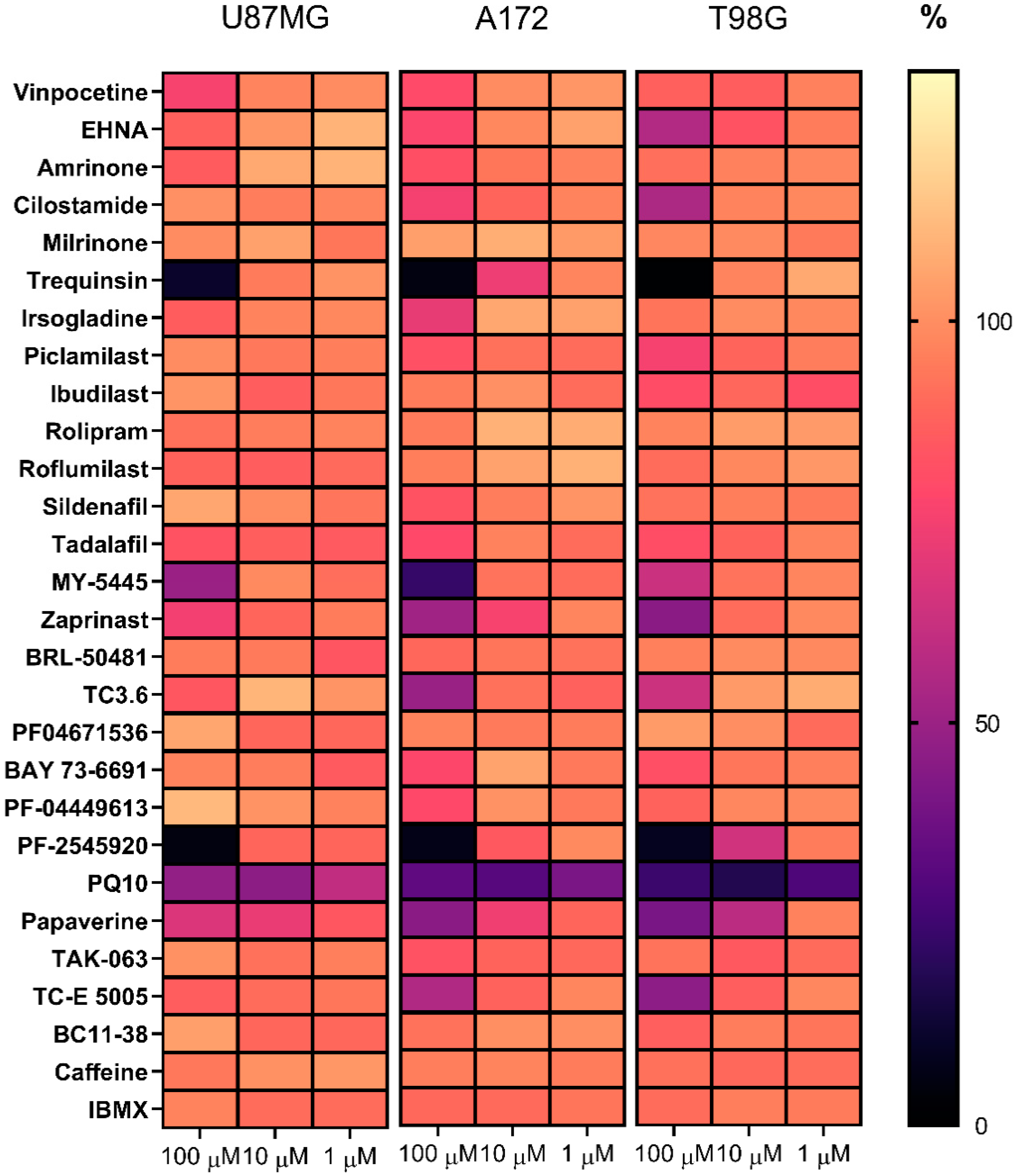{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of PDE Inhibitors on the Viability of Glioblastoma Cell Lines
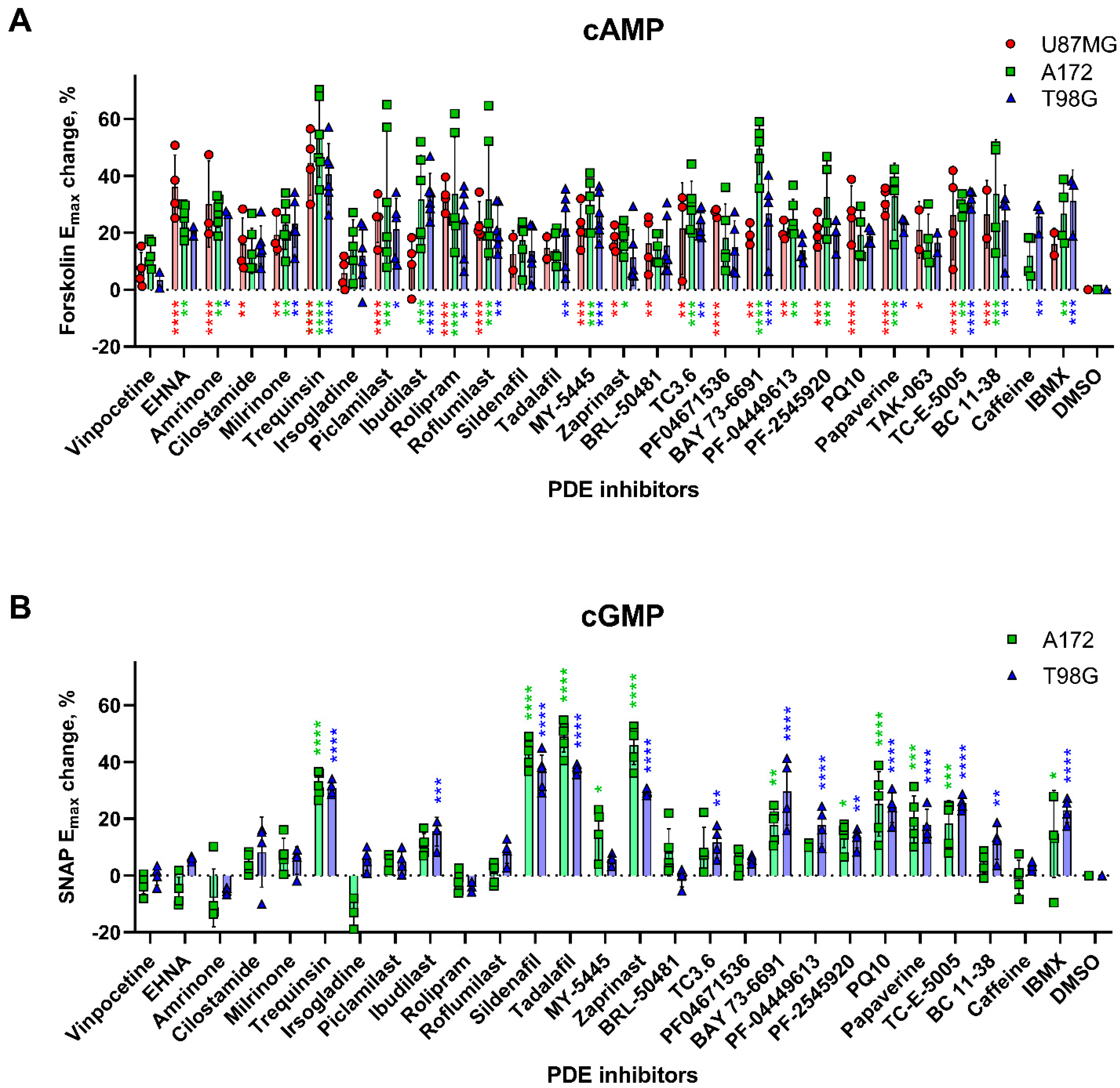
2.2. Effects of PDE Inhibitors on cAMP and cGMP Levels in Glioblastoma Cell Lines
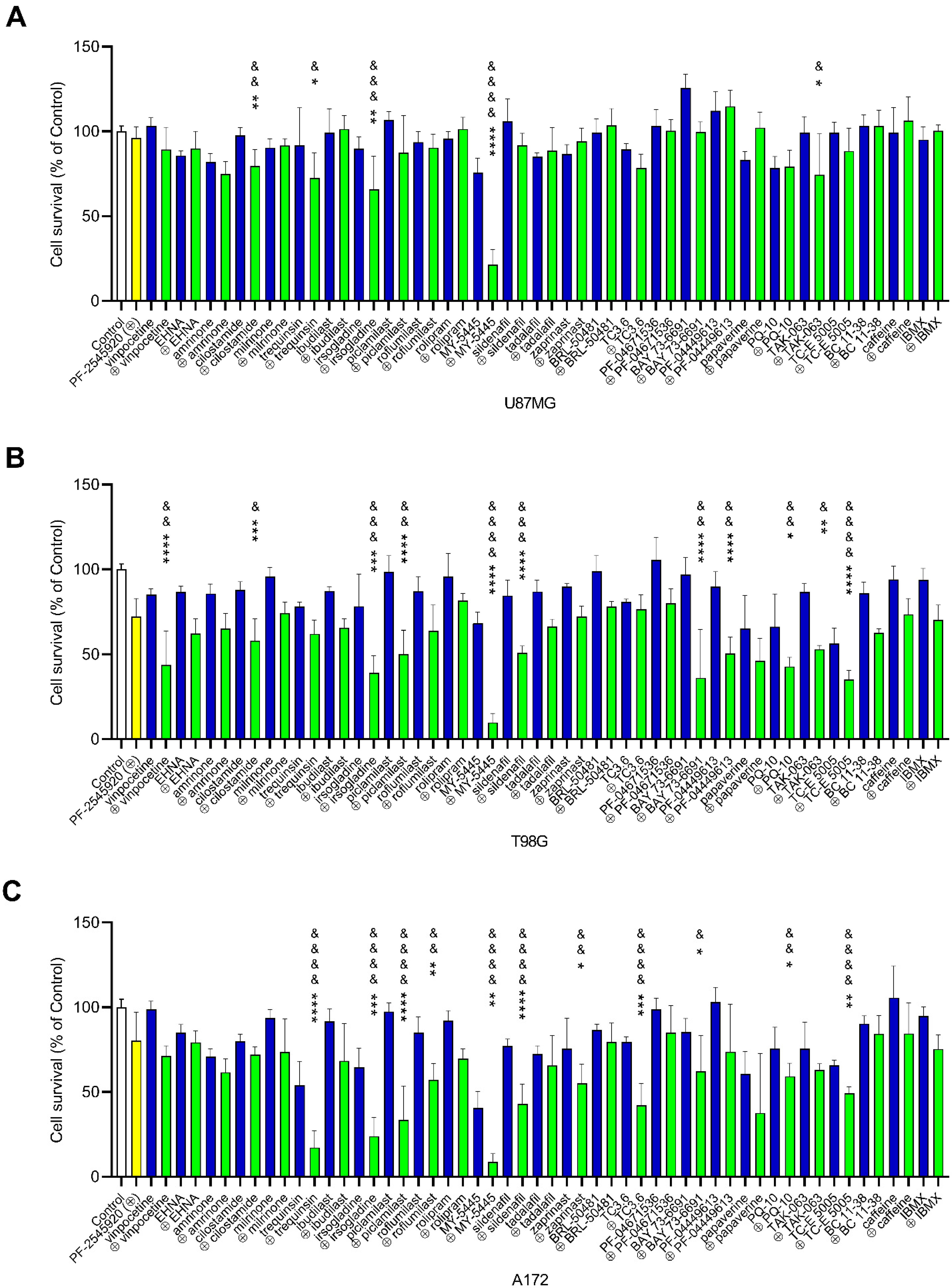
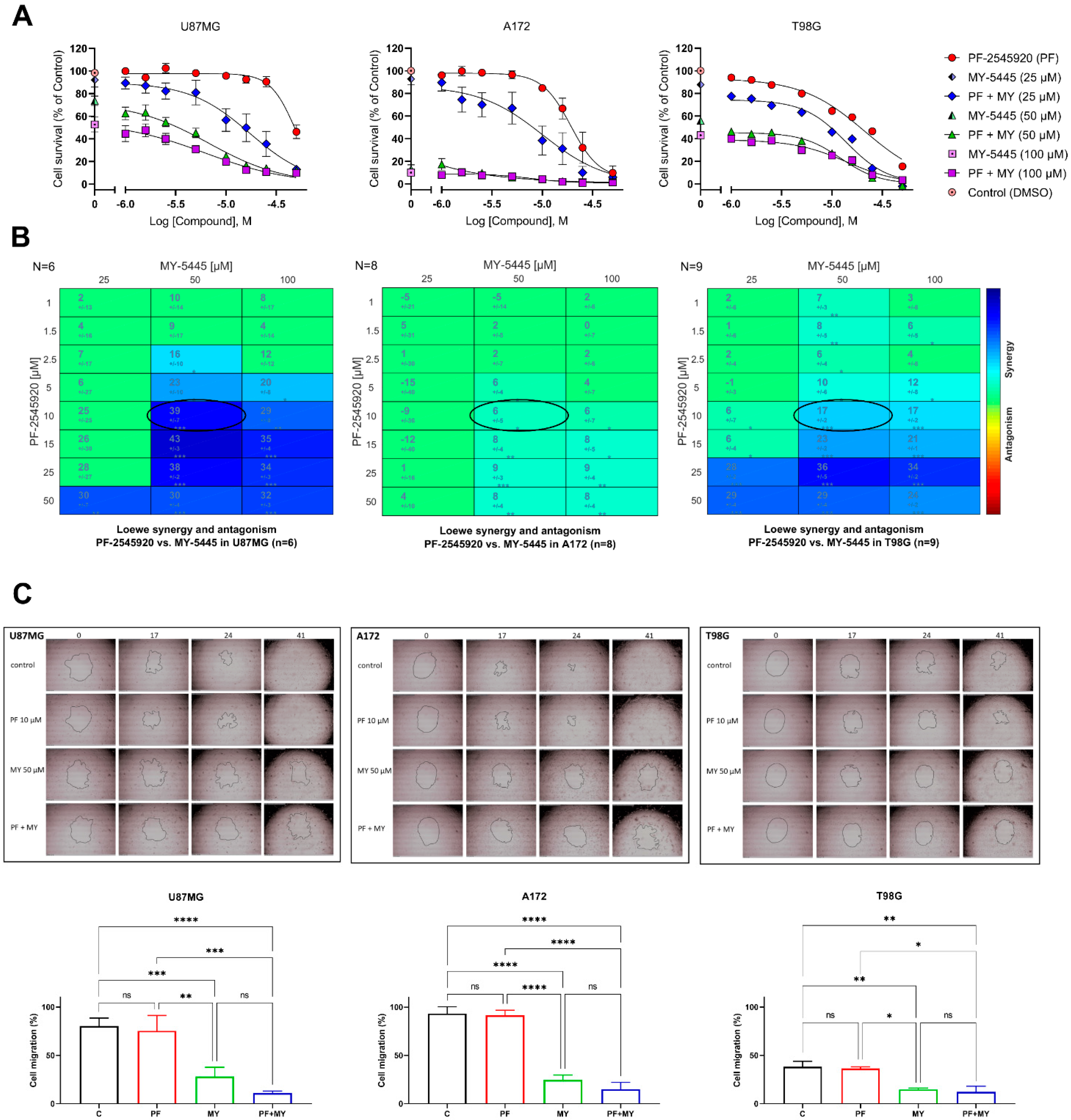
2.3. Effects of Combinations of PF-2545920 with Other PDE Inhibitors on the Survival of Glioblastoma Cells
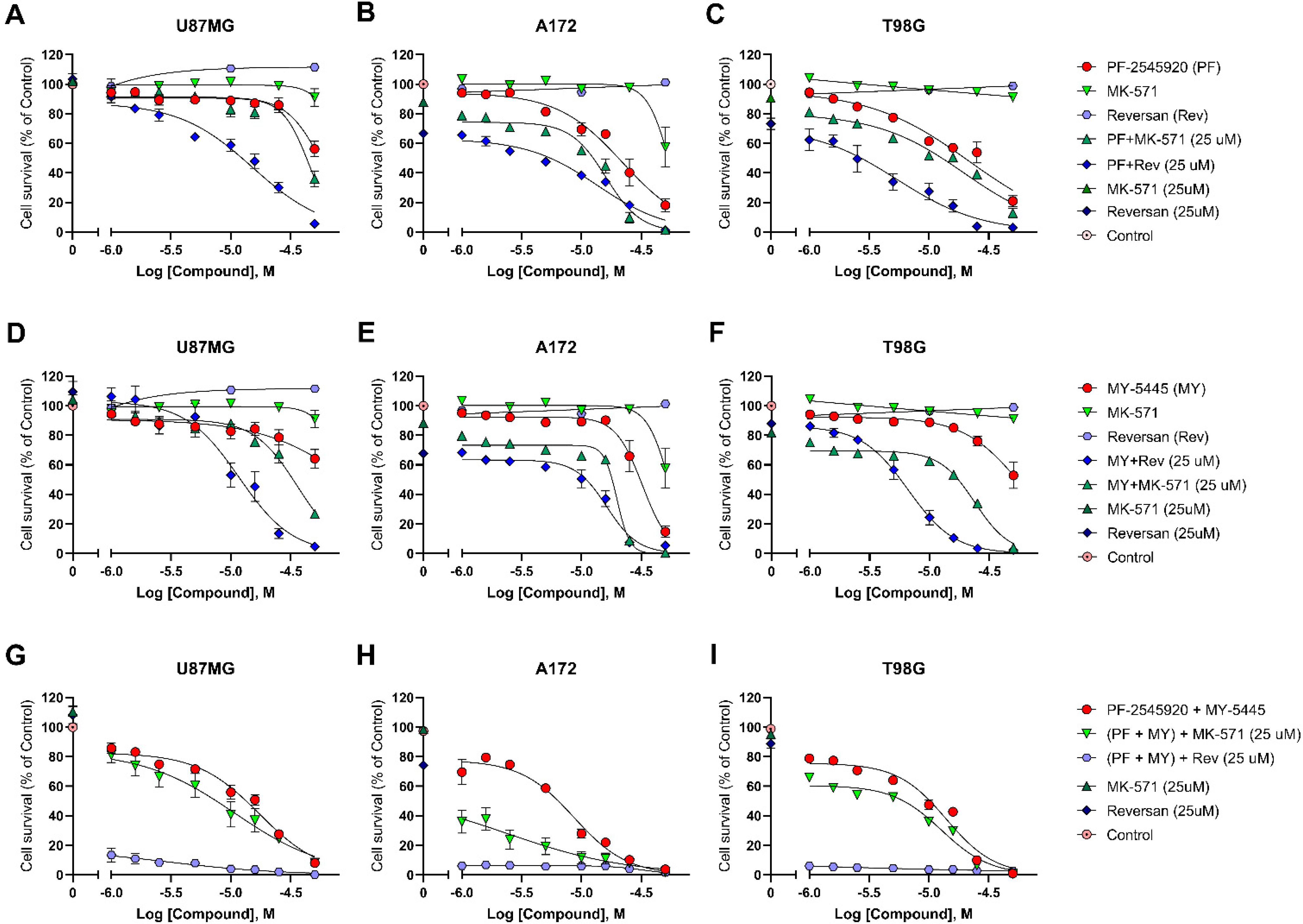
2.4. Effects of Combinations of PF-2545920 and MY-5445 with MRP1 Inhibitors on the Survival of Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. cAMP Accumulation Assay
4.4. cGMP Accumulation Assay
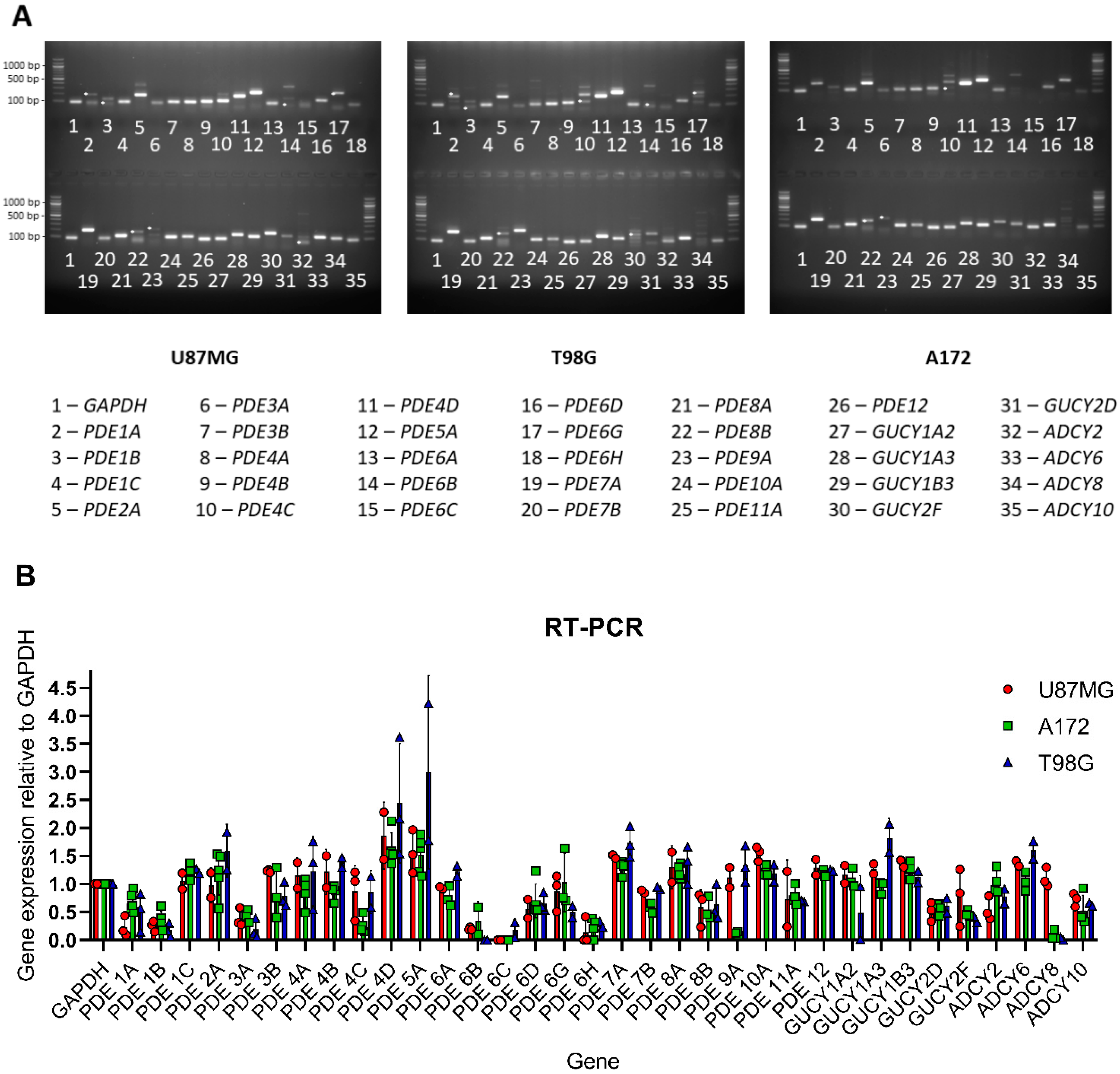
4.5. Gene Expression
4.6. Drug Combination Assays
4.7. Cell Migration Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Omuro, A.; DeAngelis, L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) Guideline on the Diagnosis and Treatment of Adult Astrocytic and Oligodendroglial Gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Brada, M.; Bent, M.J.; van den Tonn, J.-C.; Pentheroudakis, G. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in Glioblastoma: A Review on the Impact of Treatment Modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.; Svensson, F.; Kopanitsa, L.; Ladds, G.; Bailey, D. Emerging Patents in the Therapeutic Areas of Glioma and Glioblastoma. Expert Opin. Ther. Pat. 2018, 28, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for Glioblastoma: Is It Working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Savai, R.; Pullamsetti, S.S.; Banat, G.-A.; Weissmann, N.; Ghofrani, H.A.; Grimminger, F.; Schermuly, R.T. Targeting Cancer with Phosphodiesterase Inhibitors. Expert Opin. Investig. Drugs 2010, 19, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Ball, M.; Andrade, M.F.; McDermid, A.; Stanimirovic, D.B. Insulin-like Growth Factor Binding Protein-4 (IGFBP-4) Is a Novel Anti-Angiogenic and Anti-Tumorigenic Mediator Secreted by Dibutyryl Cyclic AMP (DB-cAMP)-Differentiated Glioblastoma Cells. Glia 2006, 53, 845–857. [Google Scholar] [CrossRef]
- Moon, E.-Y.; Lee, G.-H.; Lee, M.-S.; Kim, H.-M.; Lee, J.-W. Phosphodiesterase Inhibitors Control A172 Human Glioblastoma Cell Death through cAMP-Mediated Activation of Protein Kinase A and Epac1/Rap1 Pathways. Life Sci. 2012, 90, 373–380. [Google Scholar] [CrossRef]
- Kang, T.-W.; Choi, S.W.; Yang, S.-R.; Shin, T.-H.; Kim, H.-S.; Yu, K.-R.; Hong, I.-S.; Ro, S.; Cho, J.M.; Kang, K.-S. Growth Arrest and Forced Differentiation of Human Primary Glioblastoma Multiforme by a Novel Small Molecule. Sci. Rep. 2014, 4, 5546. [Google Scholar] [CrossRef]
- Daniel, P.M.; Filiz, G.; Mantamadiotis, T. Sensitivity of GBM Cells to cAMP Agonist-Mediated Apoptosis Correlates with CD44 Expression and Agonist Resistance with MAPK Signaling. Cell Death Dis. 2016, 7, e2494. [Google Scholar] [CrossRef]
- Brooks, M.D.; Jackson, E.; Warrington, N.M.; Luo, J.; Forys, J.T.; Taylor, S.; Mao, D.D.; Leonard, J.R.; Kim, A.H.; Piwnica-Worms, D.; et al. PDE7B Is a Novel, Prognostically Significant Mediator of Glioblastoma Growth Whose Expression Is Regulated by Endothelial Cells. PLoS ONE 2014, 9, e107397. [Google Scholar] [CrossRef] [PubMed]
- Rowther, F.B.; Wei, W.; Dawson, T.P.; Ashton, K.; Singh, A.; Madiesse-Timchou, M.P.; Thomas, D.G.T.; Darling, J.L.; Warr, T. Cyclic Nucleotide Phosphodiesterase-1C (PDE1C) Drives Cell Proliferation, Migration and Invasion in Glioblastoma Multiforme Cells in Vitro. Mol. Carcinog. 2016, 55, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, V.; Martini, M.; Vitiani, L.R.; Gravina, G.L.; Di Agostino, S.; Graziani, G.; D’Alessandris, Q.G.; Pallini, R.; Larocca, L.M.; Rossi, P.; et al. Type 5 Phosphodiesterase Regulates Glioblastoma Multiforme Aggressiveness and Clinical Outcome. Oncotarget 2017, 8, 13223–13239. [Google Scholar] [CrossRef]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in Neurodegenerative Disorders. IUBMB Life 2012, 64, 965–970. [Google Scholar] [CrossRef]
- Russel, F.G.M.; Koenderink, J.B.; Masereeuw, R. Multidrug Resistance Protein 4 (MRP4/ABCC4): A Versatile Efflux Transporter for Drugs and Signalling Molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef]
- Declèves, X.; Fajac, A.; Lehmann-Che, J.; Tardy, M.; Mercier, C.; Hurbain, I.; Laplanche, J.-L.; Bernaudin, J.-F.; Scherrmann, J.-M. Molecular and Functional MDR1-Pgp and MRPs Expression in Human Glioblastoma Multiforme Cell Lines. Int. J. Cancer 2002, 98, 173–180. [Google Scholar] [CrossRef]
- Benyahia, B.; Huguet, S.; Declèves, X.; Mokhtari, K.; Crinière, E.; Bernaudin, J.F.; Scherrmann, J.M.; Delattre, J.Y. Multidrug Resistance-Associated Protein MRP1 Expression in Human Gliomas: Chemosensitization to Vincristine and Etoposide by Indomethacin in Human Glioma Cell Lines Overexpressing MRP1. J. Neurooncol. 2004, 66, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Peigñan, L.; Garrido, W.; Segura, R.; Melo, R.; Rojas, D.; Cárcamo, J.G.; San Martín, R.; Quezada, C. Combined Use of Anticancer Drugs and an Inhibitor of Multiple Drug Resistance-Associated Protein-1 Increases Sensitivity and Decreases Survival of Glioblastoma Multiforme Cells in Vitro. Neurochem. Res. 2011, 36, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kögel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H.M. Inhibition of Multidrug Resistance Protein 1 (MRP1) Improves Chemotherapy Drug Response in Primary and Recurrent Glioblastoma Multiforme. Front. Neurosci. 2015, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a Novel Class of Phosphodiesterase 10A Inhibitors and Identification of Clinical Candidate 2-[4-(1-Methyl-4-Pyridin-4-Yl-1H-Pyrazol-3-Yl)-Phenoxymethyl]-Quinoline (PF-2545920) for the Treatment of Schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef]
- Tu, Z.; Fan, J.; Li, S.; Jones, L.A.; Cui, J.; Padakanti, P.K.; Xu, J.; Zeng, D.; Shoghi, K.I.; Perlmutter, J.S.; et al. Radiosynthesis and in Vivo Evaluation of [11C]MP-10 as a PET Probe for Imaging PDE10A in Rodent and Non-Human Primate Brain. Bioorg. Med. Chem. 2011, 19, 1666–1673. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Lee, K.; Xi, Y.; Zhu, B.; Gary, B.D.; Ramírez-Alcántara, V.; Gurpinar, E.; Canzoneri, J.C.; Fajardo, A.; Sigler, S.; et al. Phosphodiesterase 10A: A Novel Target for Selective Inhibition of Colon Tumor Cell Growth and β-Catenin-Dependent TCF Transcriptional Activity. Oncogene 2015, 34, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Lindsey, A.S.; Li, N.; Gary, B.; Andrews, J.; Keeton, A.B.; Piazza, G.A. β-Catenin Nuclear Translocation in Colorectal Cancer Cells Is Suppressed by PDE10A Inhibition, CGMP Elevation, and Activation of PKG. Oncotarget 2016, 7, 5353–5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- Kodama, T.; Marubayashi, T.; Kaku, M.; Mafsukado, Y. Studies on cyclic 3′,5′-AMP system in human brain and its clinical application in Neurosurgical practice (author’s transl). No Shinkei Geka 1975, 3, 43–50. [Google Scholar] [PubMed]
- Furman, M.A.; Shulman, K. Cyclic AMP and Adenyl Cyclase in Brain Tumors. J. Neurosurg. 1977, 46, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Mel’nichuk, P.V.; Shmidt, T.E.; Malakhovskiĭ, V.K.; Levchenko, L.I. Changes in the level of cyclic nucleotides in brain tumors. Zhurnal Nevropatol. Psikhiatrii Im. SS Korsakova 1978, 78, 1182–1184. [Google Scholar]
- Tsuchida, T.; Miyachi, Y.; Hayakawa, I.; Hirakawa, K.; Sano, K. Brain tumor and cyclic nucleotides. I. Concentrations of cyclic AMP and cyclic GMP in various brain tumors (author’s transl). No Shinkei 1980, 32, 59–67. [Google Scholar]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian Cyclic Nucleotide Phosphodiesterases: Molecular Mechanisms and Physiological Functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [Green Version]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic Targeting of 3′,5′-Cyclic Nucleotide Phosphodiesterases: Inhibition and Beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative Comparison of Phosphodiesterase MRNA Distribution in Human Brain and Peripheral Tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, H.-T.; O’Donnell, J.M. Phosphodiesterases in the Central Nervous System: Implications in Mood and Cognitive Disorders. Handb. Exp. Pharmacol. 2011, 204, 447–485. [Google Scholar] [CrossRef]
- Chen, T.C.; Wadsten, P.; Su, S.; Rawlinson, N.; Hofman, F.M.; Hill, C.K.; Schönthal, A.H. The Type IV Phosphodiesterase Inhibitor Rolipram Induces Expression of the Cell Cycle Inhibitors P21(Cip1) and P27(Kip1), Resulting in Growth Inhibition, Increased Differentiation, and Subsequent Apoptosis of Malignant A-172 Glioma Cells. Cancer Biol. Ther. 2002, 1, 268–276. [Google Scholar] [CrossRef]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.-I. Anticancer Effects of a Non-Narcotic Opium Alkaloid Medicine, Papaverine, in Human Glioblastoma Cells. PLoS ONE 2019, 14, e0216358. [Google Scholar] [CrossRef] [Green Version]
- Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Ryu, J.; Choi, J.; Kim, J.S.; Cho, Y.W.; Roh, G.S.; Kim, H.J.; Cho, G.J.; et al. Caffeine Inhibits Cell Proliferation and Regulates PKA/GSK3β Pathways in U87MG Human Glioma Cells. Mol. Cells 2011, 31, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Li, J.T.; Zheng, F.; Martin, E.; Kots, A.Y.; Krumenacker, J.S.; Choi, B.-K.; McCutcheon, I.E.; Weisbrodt, N.; Bögler, O.; et al. Restoring Soluble Guanylyl Cyclase Expression and Function Blocks the Aggressive Course of Glioma. Mol. Pharmacol. 2011, 80, 1076–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chou, W.-C.; Ding, Y.-M.; Wu, Y.-C. Caffeine Inhibits Migration in Glioma Cells through the ROCK-FAK Pathway. Cell. Physiol. Biochem. 2014, 33, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Miwa, S.; Hitomi, Y.; Nakamura, H.; Tsuchiya, H.; Yachie, A. Theobromine, the Primary Methylxanthine Found in Theobroma cacao, Prevents Malignant Glioblastoma Proliferation by Negatively Regulating Phosphodiesterase-4, Extracellular Signal-Regulated Kinase, Akt/Mammalian Target of Rapamycin Kinase, and Nuclear Factor-Kappa B. Nutr. Cancer 2014, 66, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, D.; Reyes-Vidal, I.; Chávez-Cortez, E.G.; Sotelo, J.; Magaña-Maldonado, R. Methylxanthines: Potential Therapeutic Agents for Glioblastoma. Pharmaceuticals 2019, 12, 130. [Google Scholar] [CrossRef] [Green Version]
- Safitri, D.; Harris, M.; Potter, H.; Yan Yeung, H.; Winfield, I.; Kopanitsa, L.; Svensson, F.; Rahman, T.; Harper, M.T.; Bailey, D.; et al. Elevated Intracellular cAMP Concentration Mediates Growth Suppression in Glioma Cells. Biochem. Pharmacol. 2020, 174, 113823. [Google Scholar] [CrossRef]
- Elhammali, A.; Ippolito, J.E.; Collins, L.; Crowley, J.; Marasa, J.; Piwnica-Worms, D. A High-Throughput Fluorimetric Assay for 2-Hydroxyglutarate Identifies Zaprinast as a Glutaminase Inhibitor. Cancer Discov. 2014, 4, 828–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benej, M.; Hong, X.; Vibhute, S.; Scott, S.; Wu, J.; Graves, E.; Le, Q.-T.; Koong, A.C.; Giaccia, A.J.; Yu, B.; et al. Papaverine and Its Derivatives Radiosensitize Solid Tumors by Inhibiting Mitochondrial Metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 10756–10761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-P.; Lusvarghi, S.; Tseng, P.-J.; Hsiao, S.-H.; Huang, Y.-H.; Hung, T.-H.; Ambudkar, S.V. MY-5445, a Phosphodiesterase Type 5 Inhibitor, Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer Drugs. Am. J. Cancer Res. 2020, 10, 164–178. [Google Scholar]
- Goldhoff, P.; Warrington, N.M.; Limbrick, D.D.; Hope, A.; Woerner, B.M.; Jackson, E.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Targeted Inhibition of Cyclic AMP Phosphodiesterase-4 Promotes Brain Tumor Regression. Clin. Cancer Res. 2008, 14, 7717–7725. [Google Scholar] [CrossRef] [Green Version]
- Inada, M.; Sato, A.; Shindo, M.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.-I. Anticancer Non-Narcotic Opium Alkaloid Papaverine Suppresses Human Glioblastoma Cell Growth. Anticancer Res. 2019, 39, 6743–6750. [Google Scholar] [CrossRef]
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast Sensitizes Glioblastoma to Temozolomide by Targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 9, 2905. [Google Scholar] [CrossRef] [Green Version]
- Sorafenib Tosylate, Valproic Acid, and Sildenafil Citrate in Treating Patients with Recurrent High-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT01817751 (accessed on 10 March 2021).
- Tamada, K.; Nakajima, S.; Ogawa, N.; Inada, M.; Shibasaki, H.; Sato, A.; Takasawa, R.; Yoshimori, A.; Suzuki, Y.; Watanabe, N.; et al. Papaverine Identified as an Inhibitor of High Mobility Group Box 1/Receptor for Advanced Glycation End-Products Interaction Suppresses High Mobility Group Box 1-Mediated Inflammatory Responses. Biochem. Biophys. Res. Commun. 2019, 511, 665–670. [Google Scholar] [CrossRef]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Singh, S.; Kim, I.-W.; Bates, S.E.; Peng, X.; Abraham, I.; Ambudkar, S.V.; et al. Sildenafil Reverses ABCB1- and ABCG2-Mediated Chemotherapeutic Drug Resistance. Cancer Res. 2011, 71, 3029–3041. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Claffey, K.P.; Brocke, S.; Epstein, P.M. Inhibition of Breast Cancer Cell Migration by Activation of cAMP Signaling. Breast Cancer Res. Treat. 2015, 152, 17–28. [Google Scholar] [CrossRef]
- Hankey, W.; Sunkel, B.; Yuan, F.; He, H.; Thomas-Ahner, J.M.; Chen, Z.; Clinton, S.K.; Huang, J.; Wang, Q. Prostate Cancer Cell Phenotypes Remain Stable Following PDE5 Inhibition in the Clinically Relevant Range. Transl. Oncol. 2020, 13, 100797. [Google Scholar] [CrossRef]
- Zimmerman, N.P.; Roy, I.; Hauser, A.D.; Wilson, J.M.; Williams, C.L.; Dwinell, M.B. Cyclic AMP Regulates the Migration and Invasion Potential of Human Pancreatic Cancer Cells. Mol. Carcinog. 2015, 54, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Hasegawa, S.; Taniguchi, K.; Yokomizo, A.; Kuwano, T.; Ono, M.; Mori, T.; Hori, S.; Kohno, K.; Kuwano, M. Possible Involvement of Multidrug-Resistance-Associated Protein (MRP) Gene Expression in Spontaneous Drug Resistance to Vincristine, Etoposide and Adriamycin in Human Glioma Cells. Int. J. Cancer 1994, 58, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Mohri, M.; Nitta, H.; Yamashita, J. Expression of Multidrug Resistance-Associated Protein (MRP) in Human Gliomas. J. Neurooncol. 2000, 49, 105–115. [Google Scholar] [CrossRef]
- Calatozzolo, C.; Gelati, M.; Ciusani, E.; Sciacca, F.L.; Pollo, B.; Cajola, L.; Marras, C.; Silvani, A.; Vitellaro-Zuccarello, L.; Croci, D.; et al. Expression of Drug Resistance Proteins Pgp, MRP1, MRP3, MRP5 and GST-Pi in Human Glioma. J. Neurooncol. 2005, 74, 113–121. [Google Scholar] [CrossRef]
- Pontén, J.; Macintyre, E.H. Long Term Culture of Normal and Neoplastic Human Glia. Acta Pathol. Microbiol. Scand. 1968, 74, 465–486. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In Vitro Cultivation of Human Tumors: Establishment of Cell Lines Derived from a Series of Solid Tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Stein, G.H. T98G: An Anchorage-Independent Human Tumor Cell Line That Exhibits Stationary Phase G1 Arrest in Vitro. J. Cell. Physiol. 1979, 99, 43–54. [Google Scholar] [CrossRef]
- canSAR BLACK—The Cancer Drug Discovery Platform. Available online: https://cansarblack.icr.ac.uk (accessed on 20 July 2021).
- Loewe, S. The Problem of Synergism and Antagonism of Combined Drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar]
- Vlot, A.H.C.; Aniceto, N.; Menden, M.P.; Ulrich-Merzenich, G.; Bender, A. Applying Synergy Metrics to Combination Screening Data: Agreements, Disagreements and Pitfalls. Drug Discov. Today 2019, 24, 2286–2298. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopanitsa, L.; Kopanitsa, M.V.; Safitri, D.; Ladds, G.; Bailey, D.S. Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition. Int. J. Mol. Sci. 2021, 22, 9665. https://doi.org/10.3390/ijms22189665
Kopanitsa L, Kopanitsa MV, Safitri D, Ladds G, Bailey DS. Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition. International Journal of Molecular Sciences. 2021; 22(18):9665. https://doi.org/10.3390/ijms22189665
Chicago/Turabian StyleKopanitsa, Liliya, Maksym V. Kopanitsa, Dewi Safitri, Graham Ladds, and David S. Bailey. 2021. "Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition" International Journal of Molecular Sciences 22, no. 18: 9665. https://doi.org/10.3390/ijms22189665
APA StyleKopanitsa, L., Kopanitsa, M. V., Safitri, D., Ladds, G., & Bailey, D. S. (2021). Suppression of Proliferation of Human Glioblastoma Cells by Combined Phosphodiesterase and Multidrug Resistance-Associated Protein 1 Inhibition. International Journal of Molecular Sciences, 22(18), 9665. https://doi.org/10.3390/ijms22189665