
Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. sRNA-Seq Analyses Revealed the Existence of 12-nt and 13-nt sRNAs
2.2. The Two Most Abundant 12-nt and 13-nt Sequences Likely Derive from 5.8S Ribosomal RNA
2.3. The C-doRNA/doRNA Ratio Differs between Species
2.4. doRNA and C-doRNA Interact with Three Proteins from the hnRNP Family
2.5. The doRNA and C-doRNA Were Broadly Localized at the Cytoplasm
2.6. C-doRNA Impairs Reporter Gene Expression Controlled by the AXIIR 5′ UTR
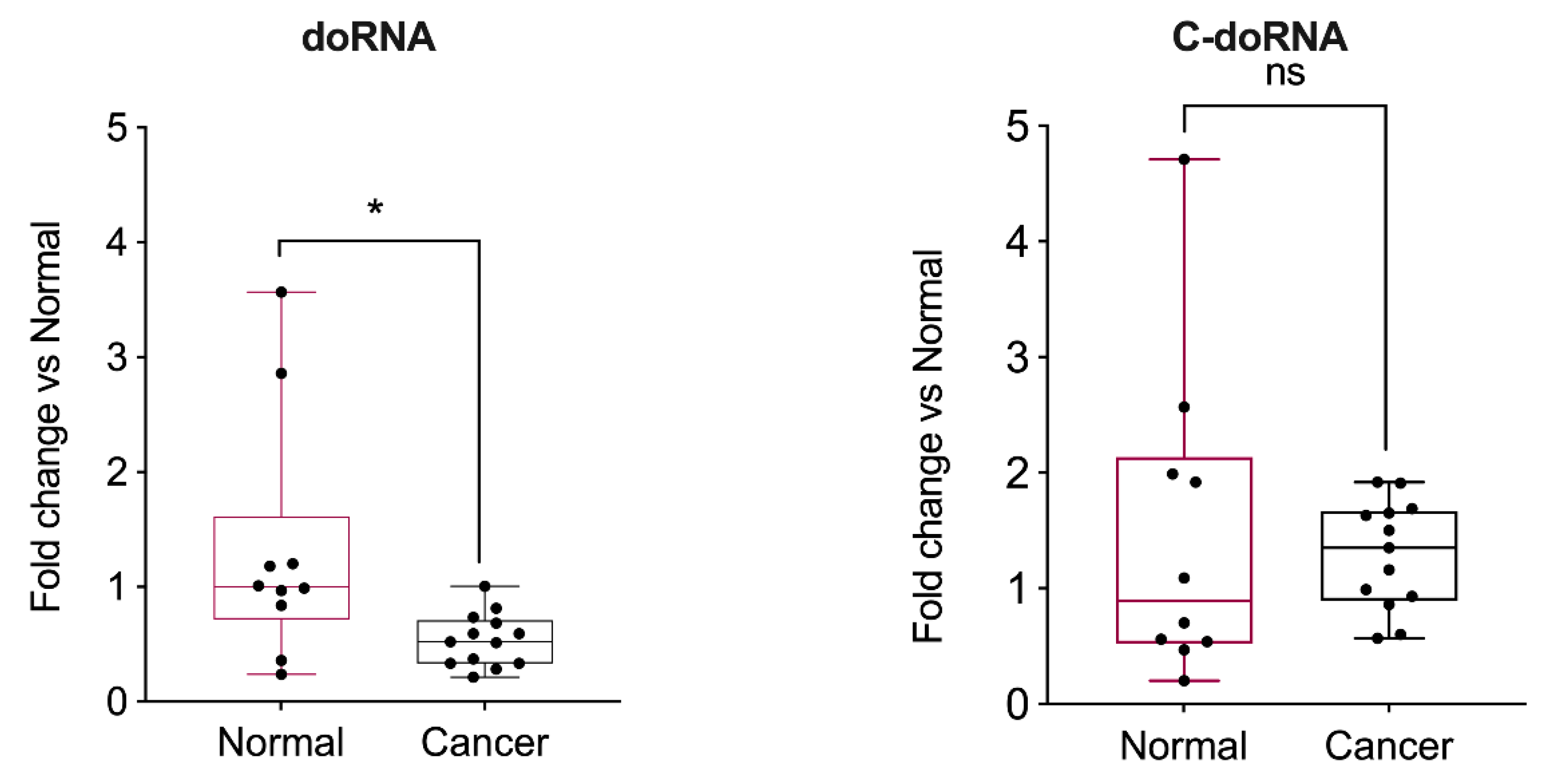
2.7. doRNA Levels, but Not C-doRNA, Are Decreased in Prostate Cancer Samples and Cell Lines
2.8. C-doRNA Reduces Cellular Migration/Proliferation of Cancer and Normal Prostate Cells
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.1.1. Human Blood Samples
4.1.2. Mouse Tissue Samples
4.1.3. Human Prostate Tissue Samples
4.2. Biological Samples
4.2.1. Primary Human Cells
4.2.2. Cultured Human Cells
4.2.3. Human Prostate Tissue Samples
4.2.4. Primary Mouse Cells and Tissues
4.2.5. Cultured Mouse Cells
4.2.6. Drosophila melanogaster
4.2.7. Arabidopsis thaliana
4.2.8. Schizosaccharomyces pombe and Saccharomyces cerevisiae
4.3. Total RNA Isolation
4.4. Small RNA Library and Sequencing
4.4.1. Library Preparation
4.4.2. sRNA-Seq
4.4.3. Bioinformatics Analysis
4.5. Adapter-Ligated RT-qPCR Method
4.6. Fractionation Cytoplasm-Nucleus Analysis
4.7. Pull-Down and Proteomics
4.8. Immunoprecipitation (IP)
4.9. Western Blot
4.10. Transfection of Fluorescent RNA and Confocal Microscopy
4.11. Dual-Luciferase Assay
4.12. Head Wound Assay (Scratch Assay)
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.E.P.; Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y. Introduction. In Computational Non-Coding RNA Biology; Zheng, Y., Ed.; Academic Press: Cambridge, MA, USA, 2019; p. 33. ISBN 978-0-12-814365-0. [Google Scholar]
- Wang, J.; Samuels, D.C.; Zhao, S.; Xiang, Y.; Zhao, Y.-Y.; Guo, Y. Current Research on Non-Coding Ribonucleic Acid (RNA). Genes 2017, 8, 366. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Delihas, N. Discovery and characterization of the first non-coding RNA that regulates gene expression, micF RNA: A historical perspective. World J. Biol. Chem. 2015, 6, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Zieve, G.; Penman, S. Small RNA species of the HeLa cell: Metabolism and subcellular localization. Cell 1976, 8, 19–31. [Google Scholar] [CrossRef]
- Mattick, J.S. Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 2003, 25, 930–939. [Google Scholar] [CrossRef]
- Churko, J.M.; Mantalas, G.L.; Snyder, M.P.; Wu, J.C. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ. Res. 2013, 112, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Uchida, Y.; Chiba, T.; Kurimoto, R.; Asahara, H. Post-transcriptional regulation of inflammation by RNA-binding proteins via cis-elements of mRNAs. J. Biochem. 2019, 166, 375–382. [Google Scholar] [CrossRef]
- Pirogov, S.A.; Gvozdev, V.A.; Klenov, M.S. Long Noncoding RNAs and Stress Response in the Nucleolus. Cells 2019, 8, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneziano, D.; Nigita, G.; Ferro, A. Computational Approaches for the Analysis of ncRNA through Deep Sequencing Techniques. Front. Bioeng. Biotechnol. 2015, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.-F.; Zhuang, Y.-Y.; Huang, F.-T.; Zhang, S.-N. Noncoding RNAs and pancreatic cancer. World J. Gastroenterol. 2016, 22, 801–814. [Google Scholar] [CrossRef]
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.; Wong, J.H.; Tsui, S.K.W.; Zuo, T.; Chan, T.F.; Ng, T.B. LncRNAs with miRNAs in regulation of gastric, liver, and colorectal cancers: Updates in recent years. Appl. Microbiol. Biotechnol. 2019, 103, 4649–4677. [Google Scholar] [CrossRef]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, G.; Veneziano, D.; Nigita, G.; Nana-Sinkam, S.P. RNA Methylation in ncRNA: Classes, Detection, and Molecular Associations. Front. Genet. 2018, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Srijyothi, L.; Saravanaraman, P.; Prathama, T.; Cheemala, A.; Baluchamy, S. Roles of Non-Coding RNAs in Transcriptional Regulation; IntechOpen: London, UK, 2018; ISBN 978-1-78923-791-7. [Google Scholar]
- Liu, D.; Mewalal, R.; Hu, R.; Tuskan, G.A.; Yang, X. New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic. Res. 2017, 4, 17031. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.-G.; Zhang, J.; Wu, H. Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Mol. Ther. Nucleic Acids 2014, 3, e161. [Google Scholar] [CrossRef]
- Vickers, K.C.; Roteta, L.A.; Hucheson-Dilks, H.; Han, L.; Guo, Y. Mining diverse small RNA species in the deep transcriptome. Trends Biochem. Sci. 2015, 40, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived from Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, V.A.; Barciszewska, M.Z.; Hochberg, A.; de Groot, N.; Barciszewski, J. Regulatory RNAs. Cell. Mol. Life Sci. 2001, 58, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Dard-Dascot, C.; Naquin, D.; d’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, A.; Gebetsberger, J.; Polacek, N. Ribosome-associated ncRNAs: An emerging class of translation regulators. RNA Biol. 2014, 11, 1335–1339. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.D.; Pagano, J.F.B.; Abdullah, F.; Ensink, W.A.; van Olst, M.; van Leeuwen, S.; Nehrdich, U.; Spaink, H.P.; Rauwerda, H.; Jonker, M.J.; et al. Identifying small RNAs derived from maternal- and somatic-type rRNAs in zebrafish development. Genome 2018, 61, 371–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S. Human 28s rRNA 5 terminal derived small RNA inhibits ribosomal protein mRNA levels. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Kim, S.W.; Lin, Y.; Moore, P.S.; Chang, Y.; John, B. Characterization of Viral and Human RNAs Smaller than Canonical MicroRNAs. J. Virol. 2009, 83, 12751–12758. [Google Scholar] [CrossRef] [Green Version]
- Plante, I.; Plé, H.; Landry, P.; Gunaratne, P.H.; Provost, P. Modulation of microRNA Activity by Semi-microRNAs. Front. Genet. 2012, 3, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: PolyA+ selection versus rRNA depletion. Sci. Rep. 2018, 8, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazar, R.N. The ribosomal 5.8S RNA: Eukaryotic adaptation or processing variant? Can. J. Biochem. Cell Biol. 1984, 62, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Awan, H.M.; Shah, A.; Rashid, F.; Wei, S.; Chen, L.; Shan, G. Comparing two approaches of miR-34a target identification, biotinylated-miRNA pulldown vs. miRNA overexpression. RNA Biol. 2018, 15, 55–61. [Google Scholar] [CrossRef]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017, 14, 1138–1152. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, L.; Guo, S.; Bu, M.; Guo, Q.; Xiong, Y.; Zhu, N.; Qiu, C.; Yan, X.; Chen, Q.; et al. hnRNPs and ELAVL1 cooperate with uORFs to inhibit protein translation. Nucleic Acids Res. 2017, 45, 2849–2864. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.-J.; Xu, X.-H.; Gao, T.; Zhang, Y.; Xia, H.; Zhao, Y.-Q.; Wang, J.-J. siRNA directed against Annexin II receptor inhibits HeLa cell proliferation, migration and invasion and induces apoptosis via suppressing ERK1/2 and Akt signaling pathways. Int. J. Clin. Exp. Pathol. 2018, 11, 88–98. [Google Scholar] [PubMed]
- D’Souza, S.; Kurihara, N.; Shiozawa, Y.; Joseph, J.; Taichman, R.; Galson, D.L.; Roodman, G.D. Annexin II interactions with the annexin II receptor enhance multiple myeloma cell adhesion and growth in the bone marrow microenvironment. Blood 2012, 119, 1888–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stage, D.E.; Eickbush, T.H. Sequence variation within the rRNA gene loci of 12 Drosophila species. Genome Res. 2007, 17, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.M.; Kurylo, C.M.; Batchelder, J.E.; Theresa Vincent, C.; Blanchard, S.C. Implications of sequence variation on the evolution of rRNA. Chromosome Res. 2019, 27, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T. Ribosomal RNA gene repeats, their stability and cellular senescence. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, M.M.; Kurylo, C.M.; Dass, R.A.; Bojmar, L.; Lyden, D.; Vincent, C.T.; Blanchard, S.C. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018, 4, eaao0665. [Google Scholar] [CrossRef] [Green Version]
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008, 18, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Grozdanov, P.; Georgiev, O.; Karagyozov, L. Complete sequence of the 45-kb mouse ribosomal DNA repeat: Analysis of the intergenic spacer. Genomics 2003, 82, 637–643. [Google Scholar] [CrossRef]
- Aubert, M.; O’Donohue, M.-F.; Lebaron, S.; Gleizes, P.-E. Pre-Ribosomal RNA Processing in Human Cells: From Mechanisms to Congenital Diseases. Biomolecules 2018, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.-F.; Chakraborty, A.; Gleizes, P.-E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef]
- Oeffinger, M.; Zenklusen, D.; Ferguson, A.; Wei, K.E.; El Hage, A.; Tollervey, D.; Chait, B.T.; Singer, R.H.; Rout, M.P. Rrp17p is a eukaryotic exonuclease required for 5′ end processing of Pre-60S ribosomal RNA. Mol. Cell 2009, 36, 768–781. [Google Scholar] [CrossRef] [Green Version]
- Henry, Y.; Wood, H.; Morrissey, J.P.; Petfalski, E.; Kearsey, S.; Tollervey, D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 1994, 13, 2452–2463. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Phillips, S.; Butler, J.S. Rat1p and Rai1p function with the nuclear exosome in the processing and degradation of rRNA precursors. RNA 2005, 11, 1571–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.W. Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol. Cell. Biol. 1997, 17, 6122–6130. [Google Scholar] [CrossRef] [Green Version]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells-focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkov, A.L. RNA Selection by PIWI Proteins. Trends Biochem. Sci. 2018, 43, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E.; Corbett, M.E.; Heeney, P.A.; Pugh, K.; Ajuh, P.M. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie 1995, 77, 22–29. [Google Scholar] [CrossRef]
- Incarnato, D.; Oliviero, S. The RNA Epistructurome: Uncovering RNA Function by Studying Structure and Post-Transcriptional Modifications. Trends Biotechnol. 2017, 35, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Nazar, R.N. Modification of rRNA as a “quality control mechanism” in ribosome biogenesis. FEBS Lett. 2002, 523, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Processing of rRNA and tRNA. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000; Section 11.6. [Google Scholar]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Sato, K.; Ishikawa, H.; Izumikawa, K.; Yamauchi, Y.; Hirota, K.; Nakayama, H.; Takahashi, N.; et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018, 46, 9289–9298. [Google Scholar] [CrossRef] [PubMed]
- Huttenhofer, A.; Kiefmann, M.; Meier-Ewert, S.; O’Brien, J.; Lehrach, H.; Bachellerie, J.P.; Brosius, J. RNomics: An experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. EMBO J. 2001, 20, 2943–2953. [Google Scholar] [CrossRef] [Green Version]
- Piekna-Przybylska, D.; Decatur, W.A.; Fournier, M.J. The 3D rRNA modification maps database: With interactive tools for ribosome analysis. Nucleic Acids Res. 2008, 36, D178–D183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matera, A.G.; Terns, R.M.; Terns, M.P. Non-coding RNAs: Lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 209–220. [Google Scholar] [CrossRef]
- Yoshida, K.; Toden, S.; Weng, W.; Shigeyasu, K.; Miyoshi, J.; Turner, J.; Nagasaka, T.; Ma, Y.; Takayama, T.; Fujiwara, T.; et al. SNORA21-An Oncogenic Small Nucleolar RNA, with a Prognostic Biomarker Potential in Human Colorectal Cancer. EBioMedicine 2017, 22, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.E.; Buffa, F.M.; Camps, C.; Ramachandran, A.; Leek, R.; Taylor, M.; Patil, M.; Sheldon, H.; Betts, G.; Homer, J.; et al. The small-nucleolar RNAs commonly used for microRNA normalisation correlate with tumour pathology and prognosis. Br. J. Cancer 2011, 104, 1168–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Xu, T.; Ganapathy, S.; Shadfan, M.; Long, M.; Huang, T.H.-M.; Thompson, I.; Yuan, Z.-M. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 2014, 33, 1348–1358. [Google Scholar] [CrossRef]
- Clark, D.P.; Pazdernik, N.J.; McGehee, M.R. Chapter 5-Manipulation of Nucleic Acids; Third, E., Ed.; Academic Cell: Cambridge, MA, USA, 2019; pp. 132–166. ISBN 978-0-12-813288-3. [Google Scholar]
- Abou Elela, S.; Nazar, R.N. Role of the 5.8S rRNA in ribosome translocation. Nucleic Acids Res. 1997, 25, 1788–1794. [Google Scholar] [CrossRef] [Green Version]
- Geissler, R.; Simkin, A.; Floss, D.; Patel, R.; Fogarty, E.A.; Scheller, J.; Grimson, A. A widespread sequence-specific mRNA decay pathway mediated by hnRNPs A1 and A2/B1. Genes Dev. 2016, 30, 1070–1085. [Google Scholar] [CrossRef] [Green Version]
- Blenkiron, C.; Hurley, D.G.; Fitzgerald, S.; Print, C.G.; Lasham, A. Links between the oncoprotein YB-1 and small non-coding RNAs in breast cancer. PLoS ONE 2013, 8, e80171. [Google Scholar] [CrossRef]
- Liu, T.T.; Arango-Argoty, G.; Li, Z.; Lin, Y.; Kim, S.W.o.; Dueck, A.; Ozsolak, F.; Monaghan, A.P.; Meister, G.; DeFranco, D.B.; et al. Noncoding RNAs that associate with YB-1 alter proliferation in prostate cancer cells. RNA 2015, 21, 1159–1172. [Google Scholar] [CrossRef] [Green Version]
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.-C.; Lee, C.H.C.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Duchez, A.-C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannee, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Levesque, T.; Rollet-Labelle, E.; et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [Green Version]
- Benmoussa, A.; Laugier, J.; Beauparlant, C.J.; Lambert, M.; Droit, A.; Provost, P. Complexity of the microRNA transcriptome of cow milk and milk-derived extracellular vesicles isolated via differential ultracentrifugation. J. Dairy Sci. 2020, 103, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Latorra, D.; Arar, K.; Hurley, J.M. Design considerations and effects of LNA in PCR primers. Mol. Cell. Probes 2003, 17, 253–259. [Google Scholar] [CrossRef]
- Astakhova, I.K.; Wengel, J. Scaffolding along nucleic acid duplexes using 2′-amino-locked nucleic acids. Acc. Chem. Res. 2014, 47, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | % Identity | Accession No. |
|---|---|---|
| Homo sapiens | ||
| Homo sapiens RNA, 5.8S ribosomal N3 (RNA5-8SN3), rRNA | 100% | NR_146153.1 |
| Homo sapiens RNA, 45S pre-ribosomal N2 (RNA45SN2), rRNA | 100% | NR_146144.1 |
| Mus musculus | ||
| Mus musculus 5.8S rRNA | 100% | K01367.1 |
| Mus musculus 18S rRNA, 5.8S rRNA and 28S rRNA | 100% | AH002077.2 |
| Drosophila melanogaster | ||
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45847), preRNA | 100% | NR_133558.1 |
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45846), preRNA | 100% | NR_133554.1 |
| Drosophila melanogaster pre-rRNA (pre-rRNA:CR45845), preRNA | 100% | NR_133549.1 |
| Drosophila melanogaster 5.8S rRNA (5.8SrRNA:CR45852) | 100% | NR_133551.1 |
| Drosophila melanogaster 5.8S and 2S rRNA | 100% | U20145.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, M.; Benmoussa, A.; Diallo, I.; Ouellet-Boutin, K.; Dorval, V.; Majeau, N.; Joly-Beauparlant, C.; Droit, A.; Bergeron, A.; Têtu, B.; et al. Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA. Int. J. Mol. Sci. 2021, 22, 9757. https://doi.org/10.3390/ijms22189757
Lambert M, Benmoussa A, Diallo I, Ouellet-Boutin K, Dorval V, Majeau N, Joly-Beauparlant C, Droit A, Bergeron A, Têtu B, et al. Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA. International Journal of Molecular Sciences. 2021; 22(18):9757. https://doi.org/10.3390/ijms22189757
Chicago/Turabian StyleLambert, Marine, Abderrahim Benmoussa, Idrissa Diallo, Katheryn Ouellet-Boutin, Véronique Dorval, Nathalie Majeau, Charles Joly-Beauparlant, Arnaud Droit, Alain Bergeron, Bernard Têtu, and et al. 2021. "Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA" International Journal of Molecular Sciences 22, no. 18: 9757. https://doi.org/10.3390/ijms22189757
APA StyleLambert, M., Benmoussa, A., Diallo, I., Ouellet-Boutin, K., Dorval, V., Majeau, N., Joly-Beauparlant, C., Droit, A., Bergeron, A., Têtu, B., Fradet, Y., Pouliot, F., & Provost, P. (2021). Identification of Abundant and Functional dodecaRNAs (doRNAs) Derived from Ribosomal RNA. International Journal of Molecular Sciences, 22(18), 9757. https://doi.org/10.3390/ijms22189757