
RNA-Seq-Based Profiling of pl Mutant Reveals Transcriptional Regulation of Anthocyanin Biosynthesis in Rice (Oryza sativa L.)



Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization
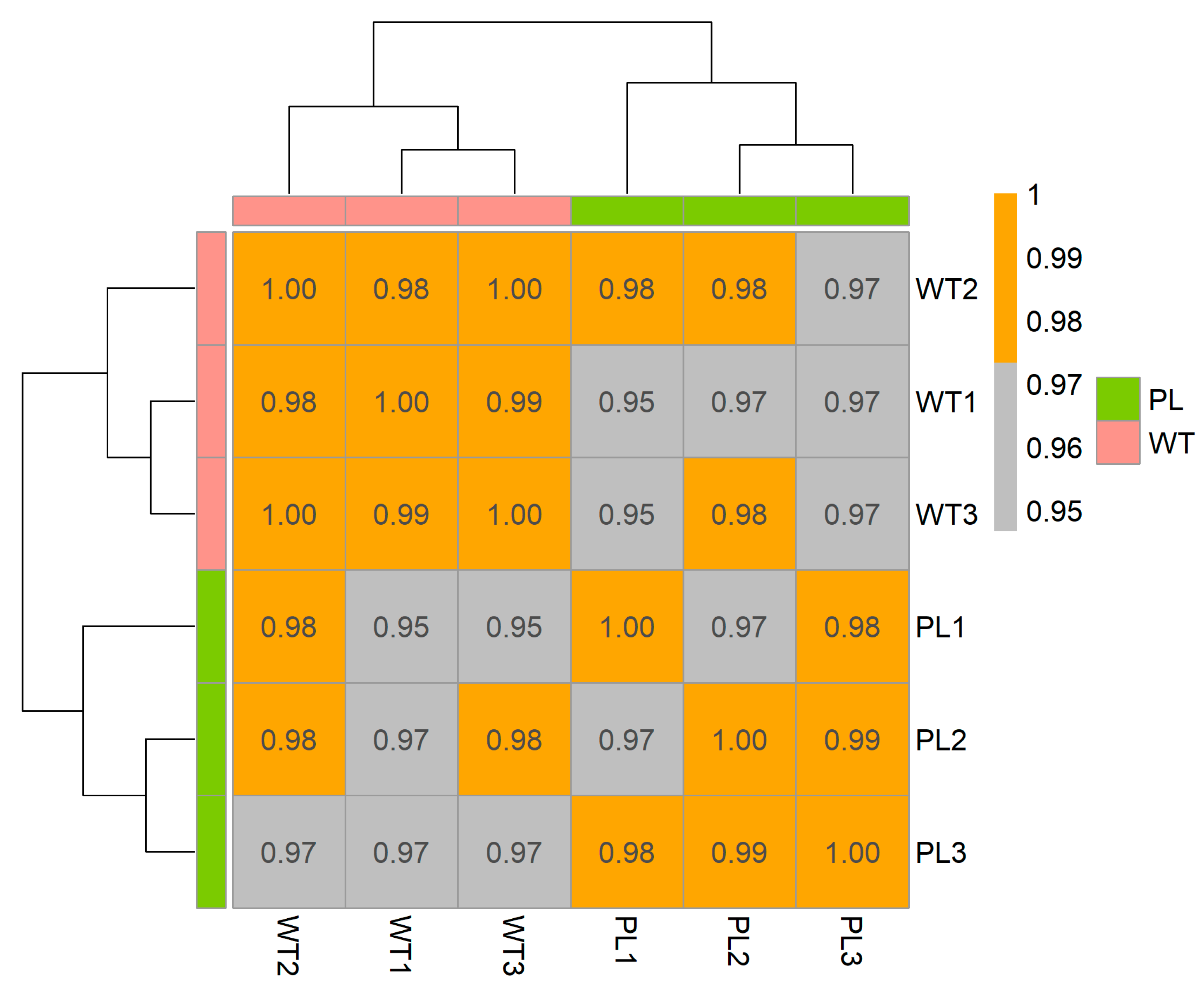
2.2. RNA-Sequence Analysis
2.3. DEGs Identification
2.4. GO Analysis
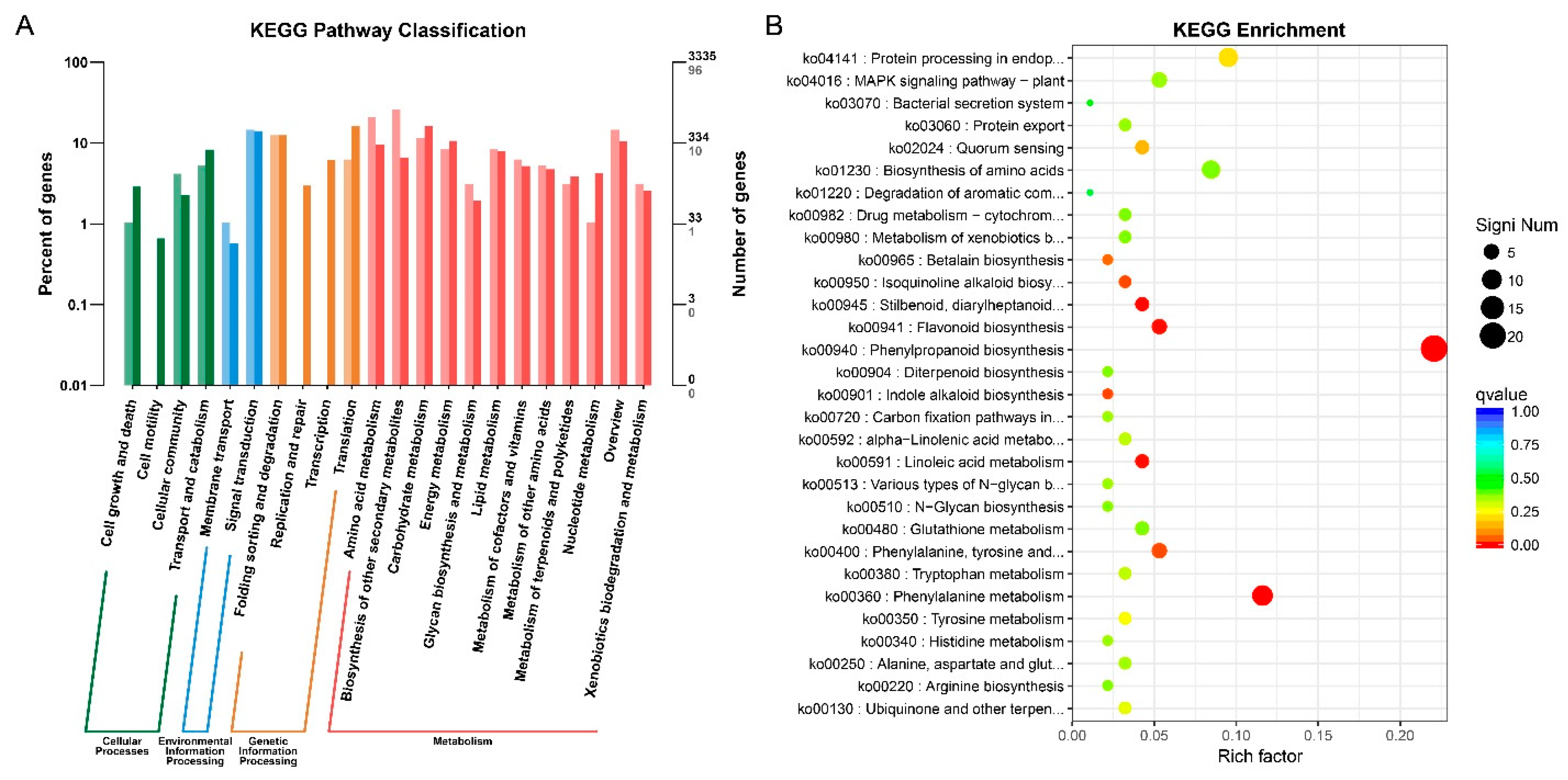
2.5. Analysis of DEGs via KEGG and KOG Pathway
2.6. Validation of the RNA-Seq Data by qRT-PCR
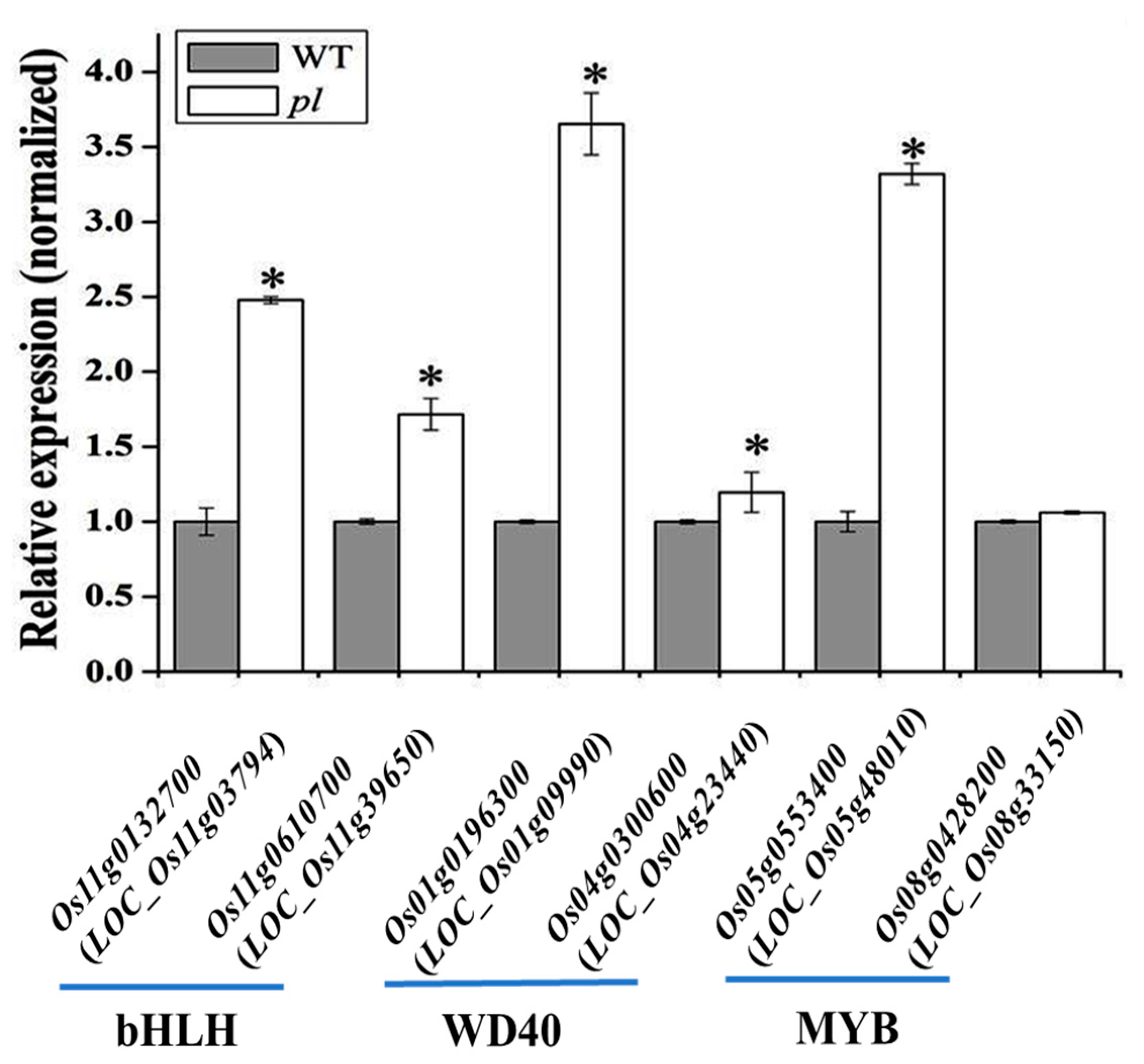
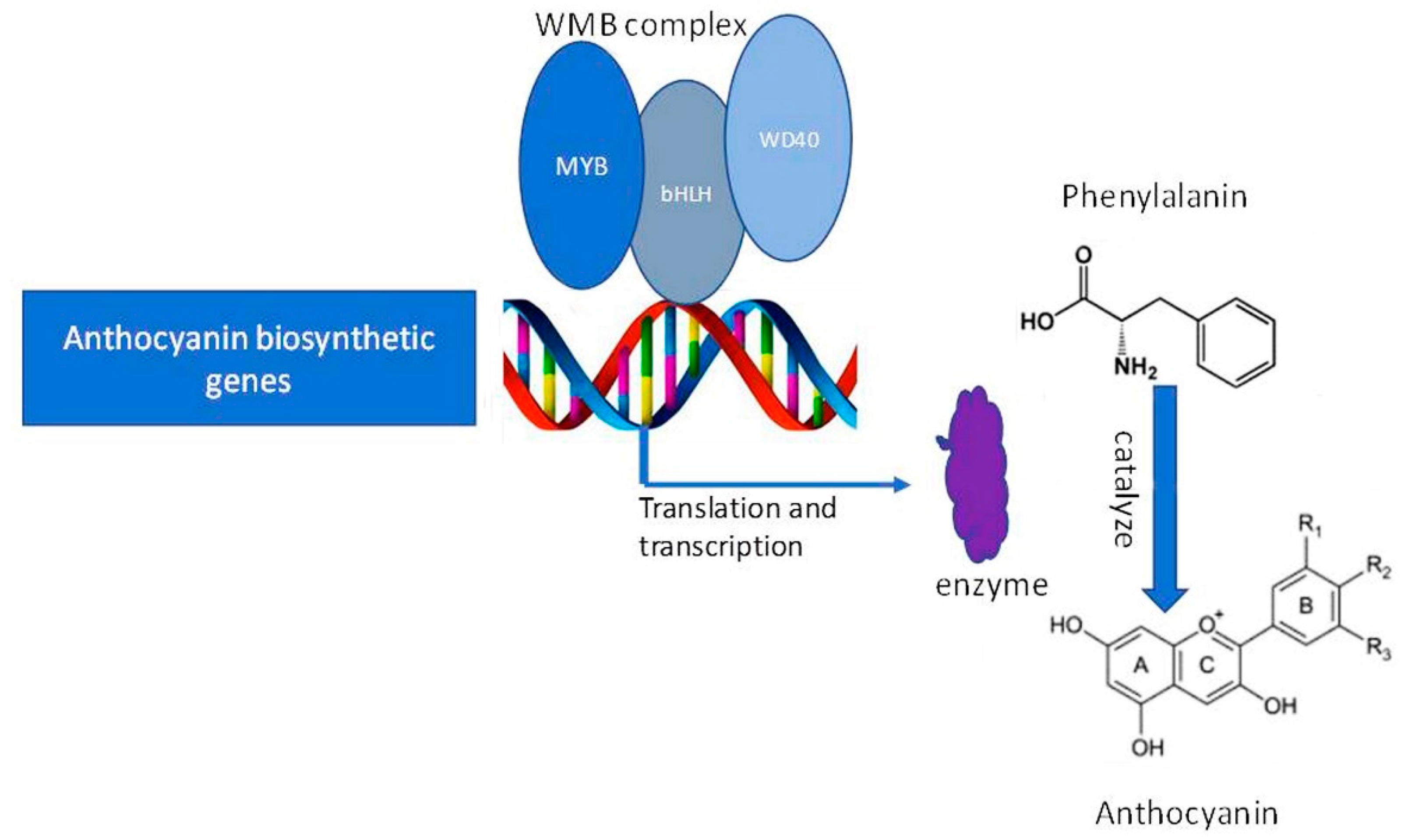
2.7. Involvement of Transcription Factors and Anthocyanin Biosynthesis in pl Mutant
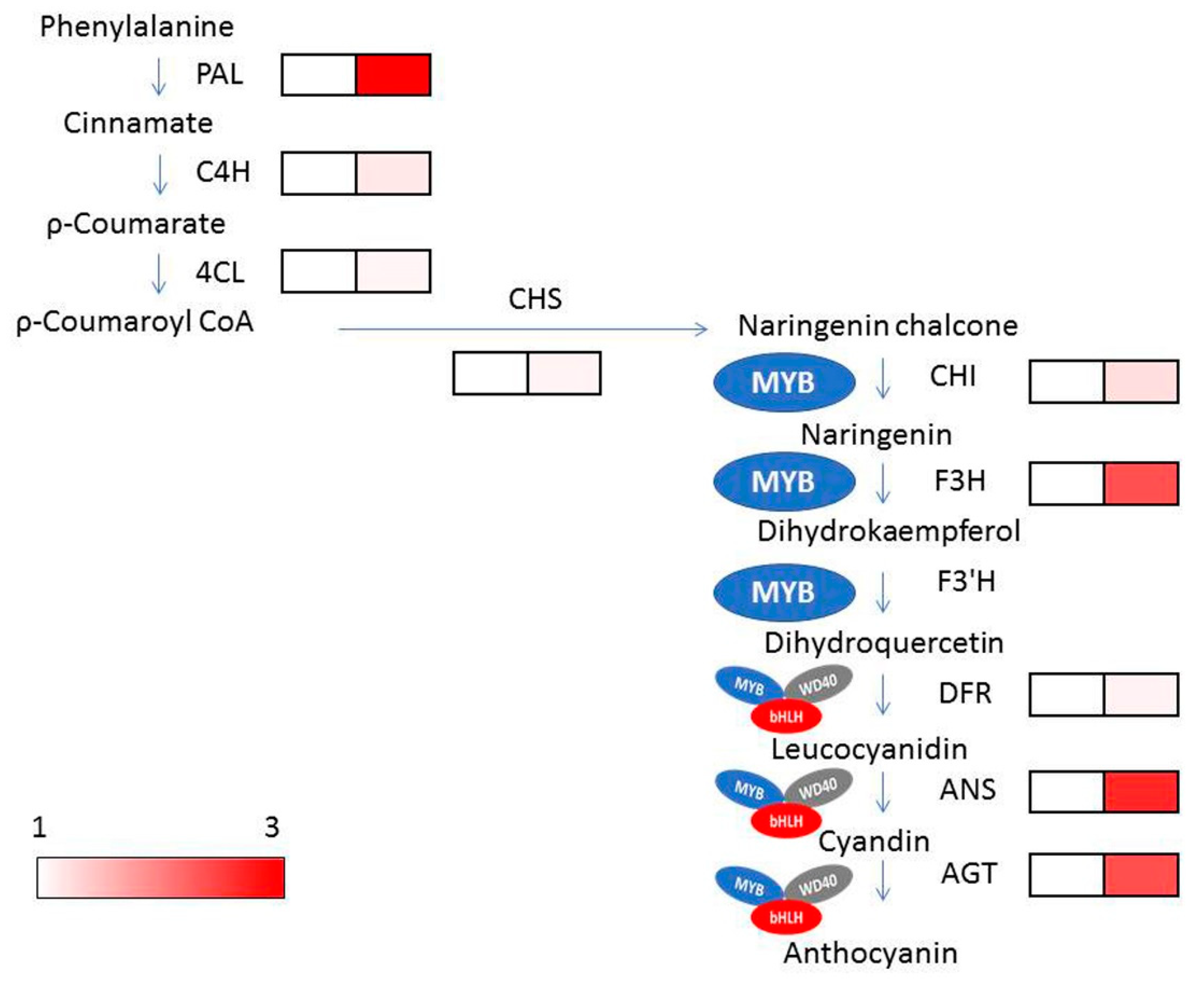
2.8. Expression Analysis of Anthocyanin Biosynthesis Genes in pl Mutant
3. Discussion
4. Materials and Experimental Methods
4.1. Plant Materials and Sample Collection
4.2. Isolation of RNA, Library Construction and Sequencing
4.3. RNA-Seq Data Assay
4.4. Transcriptomic Analysis
4.5. qRT PCR Analysis
4.6. Anthocyanin Content Measurement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Sun, X.; Wang, W.; Ding, H.; Liu, W.; Li, G.; Jiang, M.; Zhu, C.; Yao, F. Fine mapping of Pa-6 gene for purple apiculus in rice. J. Plant Biol. 2012, 55, 218–225. [Google Scholar] [CrossRef]
- Xudong, C.L.Q.Q.Z.; Zhenmin, Z.D.M.S.X. Breeding of a Photo-thermo Sensitive Genie Male Sterile Indica Rice Zhongzi S with a Purple-leaf Marker and the Hoterosis of Its Hybrid Rice Produced with It. Acta Agron. Sin. 1999, 25, 44–49. [Google Scholar]
- Zhentao, T.; Qingzhong, X. Observation on Flowering Habit of Photo—Thermoperiod Sensitive Genic Sterile Rice Lines with a Pale—Green Leaf Marker. Fen Zi Zhi Wu Yu Zhong 2004, 2, 241–245. [Google Scholar]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, H.D.; Schemske, D.W. Allele substitution at a flower colour locus produces a pollinator shift in monkeyflowers. Nature 2003, 426, 176–178. [Google Scholar] [CrossRef]
- Castellarin, S.D.; Pfeiffer, A.; Sivilotti, P.; Degan, M.; Peterlunger, E.; Di Gaspero, G. Transcriptional regulation of anthocyanin biosynthesis in ripening fruits of grapevine under seasonal water deficit. Plant Cell Environ. 2007, 30, 1381–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev-Yadun, S.; Gould, K.S. Role of Anthocyanins in Plant Defence. In Anthocyanins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 22–28. [Google Scholar] [CrossRef]
- Goswami, G.; Nath, U.K.; Park, J.I.; Hossain, M.R.; Biswas, M.K.; Kim, H.T.; Kim, H.R.; Nou, I.S. Transcriptional regulation of anthocyanin biosynthesis in a high-anthocyanin resynthesized Brassica napus cultivar. J. Biol. Res. 2018, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Z.; Zhu, M.; Zhu, Z.; Wang, Z.; Tian, S.; Chen, G. Anthocyanin accumulation and molecular analysis of correlated genes in purple kohlrabi (Brassica oleracea var. gongylodes L.). J. Agric. Food Chem. 2015, 63, 4160–4169. [Google Scholar] [CrossRef]
- Reddy, V.S.; Dash, S.; Reddy, A.R. Anthocyanin pathway in rice (Oryza sativa L.): Identification of a mutant showing dominant inhibition of anthocyanins in leaf and accumulation of proanthocyanidins in pericarp. Theor. Appl. Genet. 1995, 91, 301–312. [Google Scholar] [CrossRef]
- Petrussa, E.; Braidot, E.; Zancani, M.; Peresson, C.; Bertolini, A.; Patui, S.; Vianello, A. Plant flavonoids-biosynthesis, transport and involvement in stress responses. Int. J. Mol. Sci. 2013, 14, 14950–14973. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.P.; Skipsey, M.; Edwards, R. Roles for glutathione transferases in plant secondary metabolism. Phytochemistry 2010, 71, 338–350. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Wang, J.; Islam, F.; Li, L.; Long, M.; Yang, C.; Jin, X.; Ali, B.; Mao, B.; Zhou, W. Complementary RNA-sequencing based transcriptomics and iTRAQ proteomics reveal the mechanism of the alleviation of quinclorac stress by salicylic acid in oryza sativa ssp. japonica. Int. J. Mol. Sci. 2017, 18, 1975. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Zody, M.C. Advancing RNA-Seq analysis. Nat. Biotechnol. 2010, 28, 421–423. [Google Scholar] [CrossRef]
- Akhter, D.; Qin, R.; Nath, U.K.; Eshag, J.; Jin, X.; Shi, C. A rice gene, OsPL, encoding a MYB family transcription factor confers anthocyanin synthesis, heat stress response and hormonal signaling. Gene 2019, 699, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Wang, H.; Liu, T.; Wang, T.; Zhang, F.; Shu, X.; Zhai, H.; Wang, Z. Integrated physiological and genomic analysis reveals structural variations and expression patterns of candidate genes for colored- and green-leaf poplar. Sci. Rep. 2019, 9, 11150. [Google Scholar] [CrossRef]
- Yang, S.S.; Tu, Z.J.; Cheung, F.; Xu, W.W.; Lamb, J.A.F.S.; Jung, H.J.G.; Vance, C.P.; Gronwald, J.W. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genom. 2011, 12, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. Nrg2484-1. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Akhter, D.; Qin, R.; Nath, U.K.; Eshag, J.; Jin, X.; Shi, C. Transcriptional profile corroborates that bml mutant plays likely role in premature leaf senescence of rice (Oryza sativa L.). Int. J. Mol. Sci. 2019, 20, 1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Tan, Y.; Cui, G.; Feng, Y.; Cui, Q.; Song, X. Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, F.; Li, M.; Ma, F.; Cheng, L. Phenylpropanoid metabolites and expression of key genes involved inanthocyanin biosynthesis in the shaded peel of apple fruit in response to sun exposure. Plant Physiol. Biochem. 2013, 69, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bogs, J.; Ebadi, A.; McDavid, D.; Robinson, S.P. Identification of the flavonoid hydroxylases from grapevine and their regulation dining fruit development. Plant Physiol. 2006, 140, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.-Z.; Xie, D.-Y. Biosynthesis and Metabolic Engineering of Anthocyanins in Arabidopsis thaliana. Recent Pat. Biotechnol. 2014, 8, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, M.; Lai, Z.; Fan, B.; Shi, K.; Zhou, Y.H.; Yu, J.Q.; Chen, Z. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiol. 2010, 153, 1526–1538. [Google Scholar] [CrossRef] [Green Version]
- Rohde, A.; Morreel, K.; Ralph, J.; Goeminne, G.; Hostyn, V.; De Rycke, R.; Kushnir, S.; Van Doorsselaere, J.; Joseleau, J.P.; Vuylsteke, M.; et al. Molecular phenotyping of the pal1 and pal2 mutants of Arabidopsis thaliana reveals far-reaching consequences on phenylpropanoid, amino acid, and carbohydrate metabolism. Plant Cell 2004, 16, 2749–2771. [Google Scholar] [CrossRef] [Green Version]
- Olsen, K.M.; Lea, U.S.; Slimestad, R.; Verheul, M.; Lillo, C. Differential expression of four Arabidopsis PAL genes; PAL1 and PAL2 have functional specialization in abiotic environmental-triggered flavonoid synthesis. J. Plant Physiol. 2008, 165, 1491–1499. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.W.; Zhou, X.; Burke, S.; Wu, X.; Prior, R.L.; Li, L. The purple cauliflower arises from activation of a MYB transcription factor. Plant Physiol. 2010, 154, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Wang, Y.; Yang, S.; Xu, Y.; Chen, X. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta 2010, 232, 245–255. [Google Scholar] [CrossRef]
- Mathews, H.; Clendennen, S.K.; Caldwell, C.G.; Liu, X.L.; Connors, K.; Matheis, N.; Schuster, D.K.; Menasco, D.J.; Wagoner, W.; Lightner, J.; et al. Activation tagging in tomato identifies a transcriptional regulator of anthocyanin biosynthesis, modification, and transport. Plant Cell 2003, 15, 1689–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borevitz, J.O.; Xia, Y.; Blount, J.; Dixon, R.A.; Lamb, C. Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 2000, 12, 2383–2393. [Google Scholar] [CrossRef] [Green Version]
- An, J.P.; Zhang, X.W.; You, C.X.; Bi, S.Q.; Wang, X.F.; Hao, Y.J. MdWRKY40 promotes wounding-induced anthocyanin biosynthesis in association with MdMYB1 and undergoes MdBT2-mediated degradation. New Phytol. 2019, 224, 380–395. [Google Scholar] [CrossRef]
- He, Q.; Wu, J.; Xue, Y.; Zhao, W.; Li, R.; Zhang, L. The novel gene BrMYB2, located on chromosome A07, with a short intron 1 controls the purple-head trait of Chinese cabbage (Brassica rapa L.). Hortic. Res. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Tao, R.; Tang, Y.; Yin, L.; Ma, Y.; Ni, J.; Yan, X.; Yang, Q.; Wu, Z.; Zeng, Y.; et al. BBX16, a B-box protein, positively regulates light-induced anthocyanin accumulation by activating MYB10 in red pear. Plant Biotechnol. J. 2019, 17, 1985–1997. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Spelt, C.; Quattrocchio, F.; Mol, J.N.M.; Koes, R. Anthocyanin1 of Petunia encodes a basic helix-loop-helix protein that directly activates transcription of structural anthocyanin genes. Plant Cell 2000, 12, 1619–1631. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, J.; Hu, K.D.; Wei, S.W.; Sun, H.Y.; Hu, L.Y.; Han, Z.; Yao, G.F.; Zhang, H. PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic. Res. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- An, J.P.; Wang, X.F.; Zhang, X.W.; Xu, H.F.; Bi, S.Q.; You, C.X.; Hao, Y.J. An apple MYB transcription factor regulates cold tolerance and anthocyanin accumulation and undergoes MIEL1-mediated degradation. Plant Biotechnol. J. 2020, 18, 337–353. [Google Scholar] [CrossRef] [Green Version]
- Mano, H.; Ogasawara, F.; Sato, K.; Higo, H.; Minobe, Y. Isolation of a regulatory gene of anthocyanin biosynthesis in tuberous roots of purple-fleshed sweet potato. Plant Physiol. 2007, 143, 1252–1268. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2009, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Colin, N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.D.; Qin, R.; Li, M.; Alamin, M.; Jin, X.L.; Liu, Y.; Shi, C.H. The ferredoxin-dependent glutamate synthase (OsFd-GOGAT) participates in leaf senescence and the nitrogen remobilization in rice. Mol. Genet. Genom. 2017, 292, 385–395. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Average Raw Read Length (bp) | Clean Reads | Average Clean Read Length (bp) | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| WT | 55,160,750 | 150 | 53,875,169 | 144.14 | 0.00 | 99.20 | 97.06 | 57.83 |
| pl | 54,237,834 | 150 | 52,941,300 | 142.99 | 0.00 | 99.18 | 97.03 | 57.71 |
| Sample Name | Total Clean Reads in Mapping | Clean Read Mapped (%) | Multiple Mapped (%) | Uniquely Mapped (%) | Non-Splice Reads (%) | Splice Reads (%) | Reads Mapped in Proper Pairs (%) |
|---|---|---|---|---|---|---|---|
| WT | 45,766,956 | 84.95 | 2.74 | 82.21 | 55.13 | 27.08 | 81.31 |
| pl | 45,900,107 | 86.70 | 6.06 | 77.99 | 51.72 | 26.28 | 83.06 |
| Gene ID | Gene Name * | pl vs. WT | Gene Description | Pathway/Function Involved | |
|---|---|---|---|---|---|
| RNA-seq log2 fold change | qRT PCR fold change | ||||
| Os02g0626100 | OsPAL1 | 3.19286832 | 3.01 ± 0.01 | Similar to Phenylalanine ammonia-lyase | Anthocyanin Biosynthesis |
| Os02g0626400 | OsPAL2 | 2.10996231 | 1.93 ± 0.03 | Phenylalanine ammonia-lyase | Phenylpropanoid metabolism, Anthocyanin Biosynthesis |
| Os04g0518400 | OsPAL6 | 5.02738052 | 3.10 ± 0.19 | Similar to Phenylalanine ammonia-lyase | Phenylpropanoid metabolism, Anthocyanin Biosynthesis |
| Os05g0427400 | OsPAL7 | 3.03883343 | 1.25 ± 0.10 | Similar to Phenylalanine ammonia-lyase | Phenylpropanoid metabolism |
| Os02g0177600 | Os4CL3 | 1.82828648 | 1.19 ± 0.10 | 4-coumarate: coenzyme A ligase | Phenylpropanoid metabolism |
| Os08g0448000 | Os4CL5 | 4.87114854 | 2.08 ± 0.14 | 4-coumarate: coenzyme A ligase 5 | Phenylpropanoid metabolism |
| Os03g0122300 | OsF3H | 4.76468487 | 2.37 ± 0.24 | flavonol synthase | Flavonoid biosynthesis, Anthocyanin Biosynthesis |
| Os08g0441500 | 4.4886503 | 3.93 ± 0.015 | cinnamoyl-CoA reductase | Phenylpropanoid metabolism, Anthocyanin Biosynthesis | |
| Os05g0320700 | 1.8282864 | 1.65 ± 0.31 | cinnamate 4-hydroxylase 2 | Phenylpropanoid metabolism, Anthocyanin Biosynthesis | |
| Os10g0512400 | OsCAld5H1; F5H1 | 3.1612775 | 2.57 ± 0.15 | coniferaldehyde 5-hydroxylase 1, ferulate 5-hydroxylase 1 | Lignin biosynthesis |
| Os01g0196300 | DPF; OsbHLH025 | 5.6010349 | 3.65 ± 0.020 | basic helix-loop-helix protein 025 | |
| Os01g0838350 | Phosphate-limitation inducible gene 1 | −1.0446037 | −0.59 ± 0.09 | Conserved hypothetical protein | |
| Os01g0734800 | 1.120071 | 1.65 ± 0.10 | anthocyanidin 5,3-O-glucosyltransferase | Anthocyanin Biosynthesis | |
| Os11g0116300 | 2.70218828 | 2.25 ± 0.02 | chalcone isomerase | Anthocyanin Biosynthesis | |
| Os02g0611800 | 2.38197885 | 1.26 ± 0.08 | Hydroxycinnamoyltransferase 2 | Phenylpropanoid and Flavonoid biosynthesis | |
| Os02g0194700 | OsLOX1 | −2.9658063 | −0.68 ± 0.06 | Lipoxygenase 1 | |
| Os11g0610700 | OsWD40-190 | 1.2940596 | 1.71 ± 0.10 | WD40 repeat-like domain containing protein | |
| Os07g0543100 | −2.9658063 | −0.30 ± 0.01 | Beta-amylase 1 | ||
| Os02g0738100 | 3.24756852 | 1.19 ± 0.13 | basic helix-loop-helix protein | ||
| Os04g0662600 | 4.492093 | 1.66 ± 0.07 | Flavanone 3-hydroxylase 1 | Flavonoid biosynthesis | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.; Pan, R.; Zhang, Y.; Feng, Y.; Nath, U.K.; Gan, Y.; Shi, C.; Akhter, D. RNA-Seq-Based Profiling of pl Mutant Reveals Transcriptional Regulation of Anthocyanin Biosynthesis in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2021, 22, 9787. https://doi.org/10.3390/ijms22189787
Xu R, Pan R, Zhang Y, Feng Y, Nath UK, Gan Y, Shi C, Akhter D. RNA-Seq-Based Profiling of pl Mutant Reveals Transcriptional Regulation of Anthocyanin Biosynthesis in Rice (Oryza sativa L.). International Journal of Molecular Sciences. 2021; 22(18):9787. https://doi.org/10.3390/ijms22189787
Chicago/Turabian StyleXu, Ruonan, Ronghui Pan, Yuchan Zhang, Yanlei Feng, Ujjal Kumar Nath, Yinbo Gan, Chunhai Shi, and Delara Akhter. 2021. "RNA-Seq-Based Profiling of pl Mutant Reveals Transcriptional Regulation of Anthocyanin Biosynthesis in Rice (Oryza sativa L.)" International Journal of Molecular Sciences 22, no. 18: 9787. https://doi.org/10.3390/ijms22189787
APA StyleXu, R., Pan, R., Zhang, Y., Feng, Y., Nath, U. K., Gan, Y., Shi, C., & Akhter, D. (2021). RNA-Seq-Based Profiling of pl Mutant Reveals Transcriptional Regulation of Anthocyanin Biosynthesis in Rice (Oryza sativa L.). International Journal of Molecular Sciences, 22(18), 9787. https://doi.org/10.3390/ijms22189787