
The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
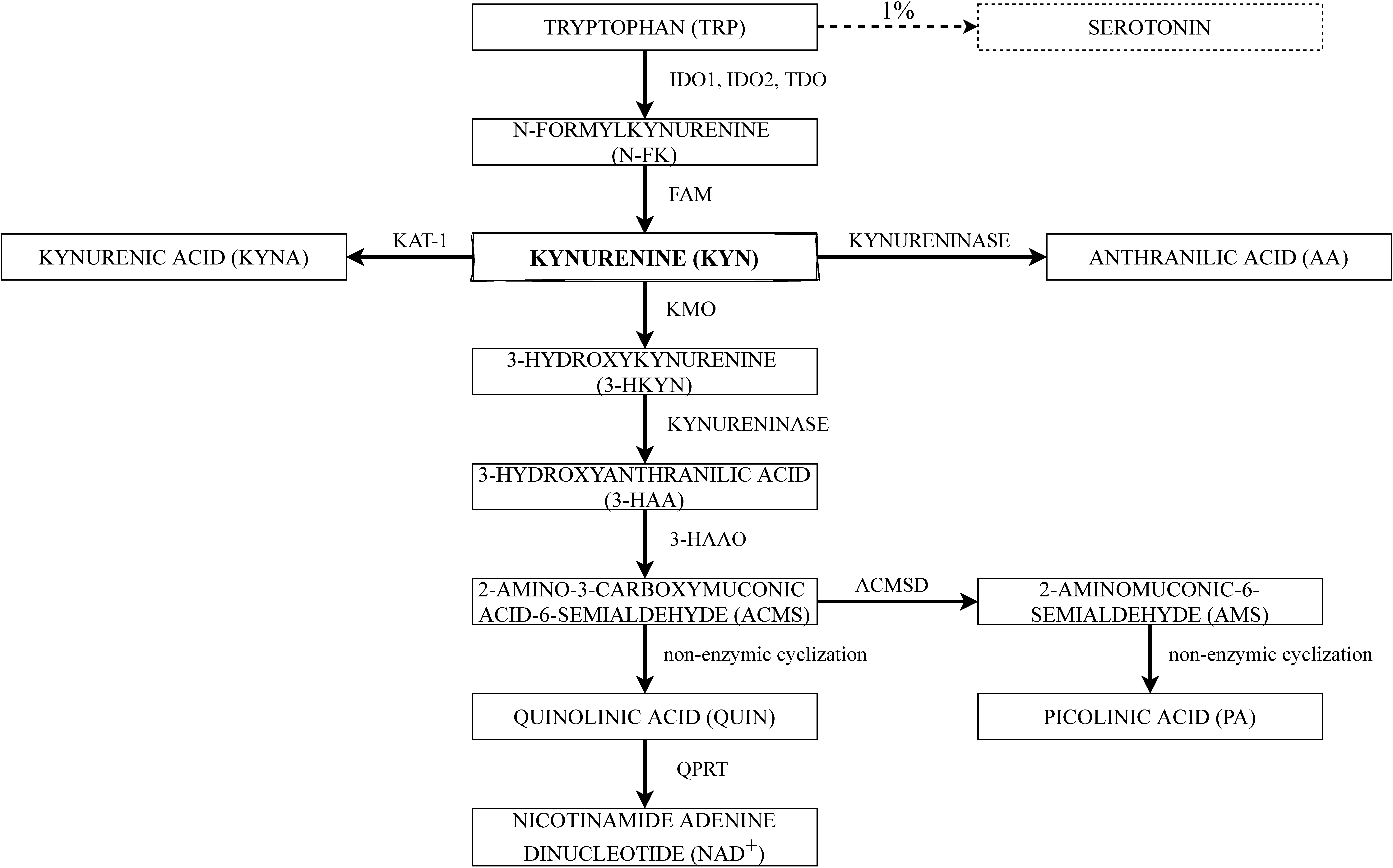
2. The Kynurenine Pathway
3. The Role of IDO1 and KP Metabolites in Immune System Regulation
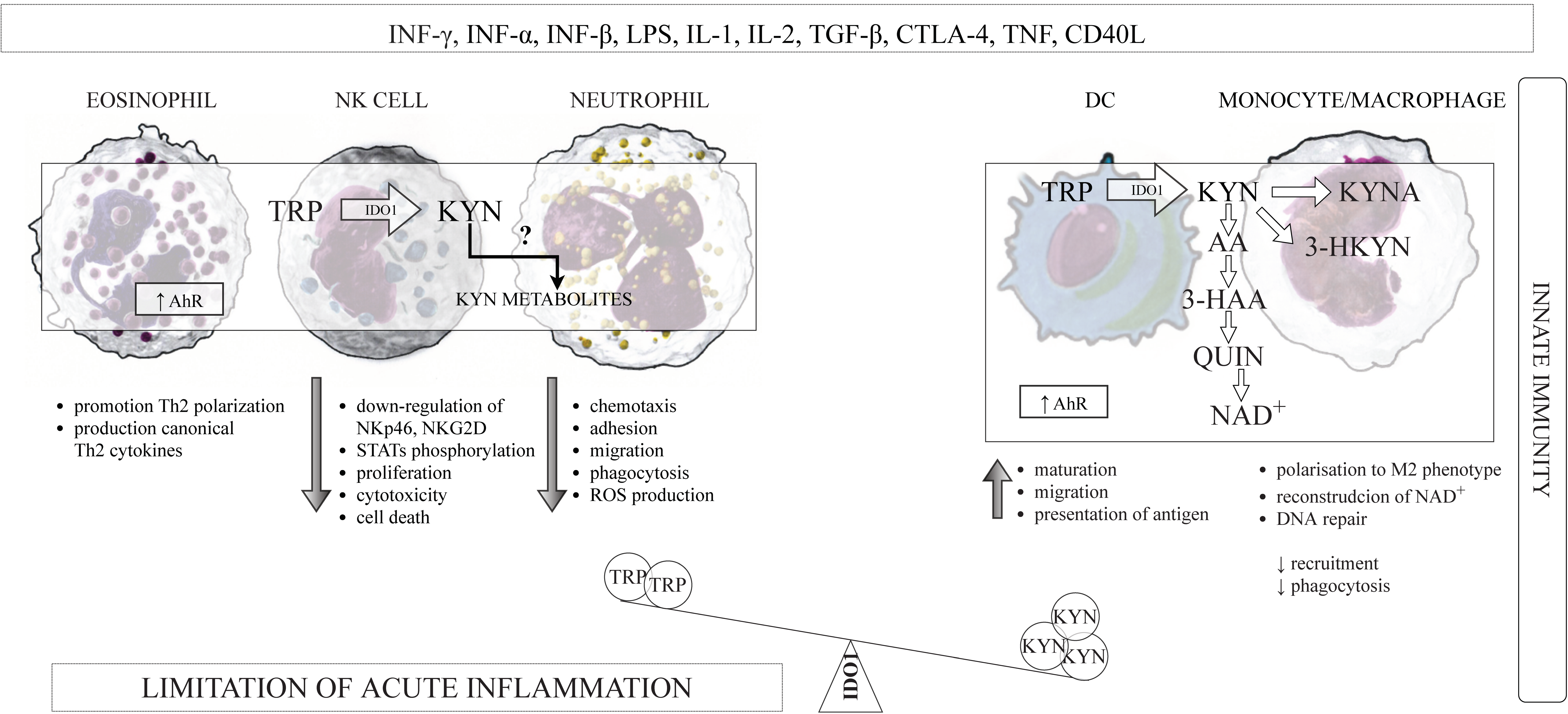
3.1. The Innate and Adaptive Immunity
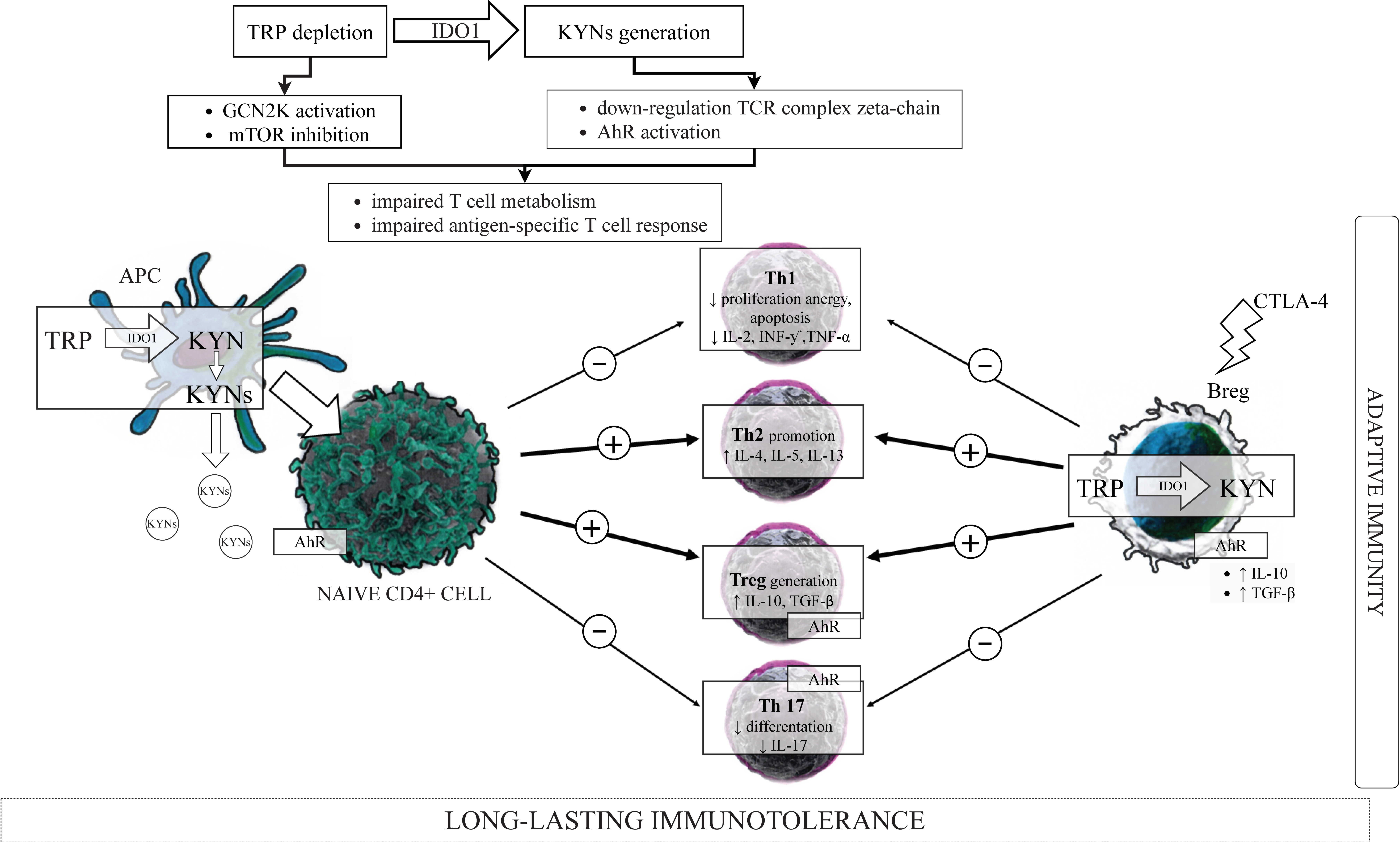
3.2. Kynurenines in the Immunoregulation—“TRP Depletion Theory” versus “TRP Utilization Theory”
3.3. Immunoregulatory Activity of IDO1
3.3.1. IDO1 and DCs
3.3.2. IDO1 and Monocytes/Macrophages
3.3.3. IDO1 and NK Cells
3.3.4. IDO1 and Eosinophiles
3.3.5. IDO1 and Neutrophils
3.4. Kynurenines and the Components of the Innate Immune System
3.5. IDO1, KYN Pathway Metabolites, and the Components of the Adaptive Immune System
3.5.1. T Cells Subsets
3.5.2. IDO1, Kynurenines and T Cells
IDO1, Kynurenines, and T Cells Metabolism
IDO1, Kynurenines, and Th1/Th2 Cells Balance
IDO1, Kynurenines and Tregs/Th17 Cells Balance
3.6. Kynurenines and IL-2 Signaling
3.7. IDO1 and B Cells
4. The Role of IDO1 and KP Activation in Autoimmunological Endocrinopathies
4.1. T1DM—An Autoimmune Disease with Unclear Pathophysiology
4.2. IDO1 and T1DM
4.3. IDO1 and Autoimmune Thyroiditis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Rose, N.R. Prediction and Prevention of Autoimmune Disease in the 21st Century: A Review and Preview. Am. J. Epidemiol. 2016, 183, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, R.M.; Giuffrida, G.; Campennì, A. Autoimmune endocrine diseases. Min. Endocrinol. 2018, 43, 305–322. [Google Scholar]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef]
- Iddah, M.A.; Macharia, B.N. Autoimmune thyroid disorders. ISRN Endocrinol. 2013, 2013, 509764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.J.; Hegedüs, L. Graves’ disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, B.; Pearce, S.H. Diagnosis and management of thyrotoxicosis. BMJ 2014, 349, g5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, A.; Fallahi, P.; Elia, G.; Ragusa, F.; Paparo, S.R.; Ruffilli, I.; Patrizio, A.; Gonnella, D.; Giusti, C.; Virili, C.; et al. Graves’ disease: Clinical manifestations, immune pathogenesis (cytokines and chemokines) and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101388. [Google Scholar] [CrossRef] [PubMed]
- Altobelli, E.; Petrocelli, R.; Verrotti, A.; Chiarelli, F.; Marziliano, C. Genetic and environmental factors affect the onset of type 1 diabetes mellitus. Pediatr. Diabetes 2016, 17, 559–566. [Google Scholar] [CrossRef]
- Rogers, M.A.M.; Kim, C.; Banerjee, T.; Lee, J.M. Fluctuations in the incidence of type 1 diabetes in the United States from 2001 to 2015: A longitudinal study. BMC Med. 2017, 15, 199. [Google Scholar] [CrossRef] [PubMed]
- Krischer, J.P.; Lynch, K.F.; Schatz, D.A.; Ilonen, J.; Lernmark, Å.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; Simell, O.G.; Toppari, J.; et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: The TEDDY study. Diabetologia 2015, 58, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013, 309, 2473–2479. [Google Scholar] [CrossRef] [Green Version]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Cutolo, M. Autoimmune polyendocrine syndromes. Autoimmun. Rev. 2014, 13, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Ben-Skowronek, I.; Michalczyk, A.; Piekarski, R.; Wysocka-Łukasik, B.; Banecka, B. Type III Polyglandular Autoimmune Syndromes in children with type 1 diabetes mellitus. Ann. Agric. Environ. Med. 2013, 20, 140–146. [Google Scholar] [PubMed]
- Betterle, C.; Presotto, F.; Furmaniak, J. Epidemiology, pathogenesis, and diagnosis of Addison’s disease in adults. J. Endocrinol. Investig. 2019, 42, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Schön, M.P. Adaptive and Innate Immunity in Psoriasis and Other Inflammatory Disorders. Front. Immunol. 2019, 10, 1764. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Lu, M.P.; Wang, J.H.; Xu, M.; Yang, S.R. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J. Pediatr. 2020, 16, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun. Rev. 2018, 17, 142–154. [Google Scholar] [CrossRef]
- Wahren-Herlenius, M.; Dörner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Elia, G.; Ragusa, F.; Ruffilli, I.; Paparo, S.R.; Antonelli, A. Thyroid autoimmune disorders and cancer. Semin. Cancer Biol. 2020, 64, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xi, S.; He, G.; Li, Z.; Gang, X.; Sun, C.; Guo, W.; Wang, G. Two to Tango: Dialogue between Adaptive and Innate Immunity in Type 1 Diabetes. J. Diabetes Res. 2020, 2020, 4106518. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Routy, B.; Graziani, G.M.; Mehraj, V. The Kynurenine Pathway Is a Double-Edged Sword in Immune-Privileged Sites and in Cancer: Implications for Immunotherapy. Int. J. Tryptophan Res. 2016, 9, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Varo, N.; Alegre, E.; Díaz, A.; Melero, I. Immunosuppression routed via the kynurenine pathway: A biochemical and pathophysiologic approach. Adv. Clin. Chem. 2008, 45, 155–197. [Google Scholar]
- Sorgdrager, F.J.H.; Naudé, P.J.W.; Kema, I.P.; Nollen, E.A.; Deyn, P.P. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front. Immunol. 2019, 10, 2565. [Google Scholar] [CrossRef]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural. Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Bo, L.; Guojun, T.; Li, G. An Expanded Neuroimmunomodulation Axis: sCD83-Indoleamine 2,3-Dioxygenase-Kynurenine Pathway and Updates of Kynurenine Pathway in Neurologic Diseases. Front. Immunol. 2018, 9, 1363. [Google Scholar] [CrossRef]
- Mondanelli, G.; Iacono, A.; Carvalho, A.; Orabona, C.; Volpi, C.; Pallotta, M.T.; Matino, D.; Esposito, S.; Grohmann, U. Amino acid metabolism as drug target in autoimmune diseases. Autoimmun. Rev. 2019, 18, 334–348. [Google Scholar] [CrossRef]
- Badawy, A.A. Tryptophan availability for kynurenine pathway metabolism across the life span: Control mechanisms and focus on aging, exercise, diet and nutritional supplements. Neuropharmacology 2017, 112 Pt B, 248–263. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Fernstrom, J.D. Branched-chain amino acids and brain function. J. Nutr. 2005, 135 (Suppl. S6), 1539S–1546S. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanain, H.H.; Chon, S.Y.; Gupta, S.L. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J. Biol. Chem. 1993, 268, 5077–5084. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Orabona, C.; Grohmann, U.; Puccetti, P. TGFbeta and kynurenines as the key to infectious tolerance. Trends Mol. Med. 2009, 15, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chaves, A.C.; Cerávolo, I.P.; Gomes, J.A.; Zani, C.L.; Romanha, A.J.; Gazzinelli, R.T. IL-4 and IL-13 regulate the induction of indoleamine 2,3-dioxygenase activity and the control of Toxoplasma gondii replication in human fibroblasts activated with IFN-gamma. Eur. J. Immunol. 2001, 31, 333–344. [Google Scholar] [CrossRef]
- Yadav, M.C.; Burudi, E.M.; Alirezaei, M.; Flynn, C.C.; Watry, D.D.; Lanigan, C.M.; Fox, H.S. IFN-gamma-induced IDO and WRS expression in microglia is differentially regulated by IL-4. Glia 2007, 55, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuasa, H.J.; Takubo, M.; Takahashi, A.; Hasegawa, T.; Noma, H.; Suzuki, T. Evolution of vertebrate indoleamine 2,3-dioxygenases. J. Mol. Evol. 2007, 65, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan—kynurenine metabolism. Ann. N. Y. Acad. Sci. 2010, 1199, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Haber, R.; Bessette, D.; Hulihan-Giblin, B.; Durcan, M.J.; Goldman, D. Identification of tryptophan 2,3-dioxygenase RNA in rodent brain. J. Neurochem. 1993, 60, 1159–1162. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Takeuchi, F.; Shibata, Y. Purification of L-kynurenine 3-hydroxylase by affinity chromatography. J. Chromatogr. 1979, 169, 357–364. [Google Scholar] [CrossRef]
- Tanizawa, K.; Soda, K. Inducible and constitutive kynureninases. Control of the inducible enzyme activity by transamination and inhibition of the constitutive enzyme by 3-hydroxyanthranilate. J. Biochem. 1979, 86, 499–508. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Stone, T.W. Development and therapeutic potential of kynurenic acid and kynurenine derivatives for neuroprotection. Trends Pharmacol. Sci. 2000, 21, 149–154. [Google Scholar] [CrossRef]
- Clark, R.; Kupper, T. Old meets new: The interaction between innate and adaptive immunity. J. Investig. Dermatol. 2005, 125, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Riera Romo, M.; Perez-Martinez, D.; Castillo Ferrer, C. Innate immunity in vertebrates: An overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.R.; Ozaki, Y.; Datta, S.P.; Borden, E.C.; Sondel, P.M.; Malone, D.G. Implications of interferon-induced tryptophan catabolism in cancer, auto-immune diseases and AIDS. Adv. Exp. Med. Biol. 1991, 294, 425–435. [Google Scholar]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Jiga, L.P.; Chuang, J.J.; Randazzo, M.; Opelz, G.; Terness, P. Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: Tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl. Int. 2005, 18, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Harden, J.L.; Egilmez, N.K. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol. Investig. 2012, 41, 738–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.F.; Wang, H.S.; Wang, H.; Zhang, F.; Wang, K.F.; Guo, Q.; Zhang, G.; Cai, S.H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Odemuyiwa, S.O.; Ghahary, A.; Li, Y.; Puttagunta, L.; Lee, J.E.; Musat-Marcu, S.; Ghahary, A.; Moqbel, R. Cutting edge: Human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J. Immunol. 2004, 173, 5909–5913. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Zarember, K.A.; Long-Priel, D.; Chan, K.C.; Fox, S.D.; Gallin, J.I.; Kuhns, D.B.; Malech, H.L. Tryptophan/kynurenine metabolism in human leukocytes is independent of superoxide and is fully maintained in chronic granulomatous disease. Blood 2010, 116, 1755–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, S.; Goto, S.; Tahara, K.; Sasaki, A.; Kawano, K.; Kitano, S. Inhibition of indoleamine 2,3-dioxygenase suppresses NK cell activity and accelerates tumor growth. J. Exp. Ther. Oncol. 2003, 3, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Nouël, A.; Pochard, P.; Simon, Q.; Ségalen, I.; Le Meur, Y.; Pers, J.O.; Hillion, S. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J. Autoimmun. 2015, 59, 53–60. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Kadowaki, N.; Antonenko, S.; Lau, J.Y.; Liu, Y.J. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 2000, 192, 219–226. [Google Scholar] [CrossRef]
- Young, L.J.; Wilson, N.S.; Schnorrer, P.; Proietto, A.; Ten Broeke, T.; Matsuki, Y.; Mount, A.M.; Belz, G.T.; O’Keeffe, M.; Ohmura-Hoshino, M.; et al. Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat. Immunol. 2008, 9, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. TH17 cells in development: An updated view of their molecular identity and genetic programming. Nat. Rev. Immunol. 2008, 8, 337–348. [Google Scholar] [CrossRef]
- Blum, A.; Chaperot, L.; Molens, J.P.; Foissaud, V.; Plantaz, D.; Plumas, J. Mechanisms of TRAIL-induced apoptosis in leukemic plasmacytoid dendritic cells. Exp. Hematol. 2006, 34, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Vermi, W.; Cavani, A.; Lonardi, S.; Carbone, T.; Facchetti, F.; Bosisio, D.; Sozzani, S. IL-21 May Promote Granzyme B-Dependent NK/Plasmacytoid Dendritic Cell Functional Interaction in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow-Anacker, A.; Bochkov, Y.; Gern, J.; Seroogy, C.M. Neonatal immune response to rhinovirus A16 has diminished dendritic cel function and increased B cell activation. PLoS ONE 2017, 12, e0180664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallarino, F.; Vacca, C.; Orabona, C.; Belladonna, M.L.; Bianchi, R.; Marshall, B.; Keskin, D.B.; Mellor, A.L.; Fioretti, M.C.; Grohmann, U.; et al. Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int. Immunol. 2002, 14, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Reizis, B.; Colonna, M.; Trinchieri, G.; Barat, F.; Gilliet, M. Plasmacytoid dendritic cells: One-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 2011, 11, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Daissormont, I.T.; Christ, A.; Temmerman, L.; Sampedro Millares, S.; Seijkens, T.; Rousch, M.; Poggi, M.; Boon, L.; Van Der Loos, C.; Daemen, M.; et al. Plasmacytoid dendritic cells protect against atheroslerosis by tuning T-cell proliferation and activity. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO activates regulatory T cells and block their conversion into Th17-like T cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef]
- Mellor, A.L.; Chandler, P.; Baban, B.; Hansen, A.M.; Marshall, B.; Pihkala, J.; Waldmann, H.; Cobbold, S.; Adams, E.; Munn, D.H. Specific subsets of murine dendritic cells acquire potent T-cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3-dioxygenase. Int. Immunol. 2004, 16, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Chandler, P.R.; Johnson, B.A.; Huang, L.; Li, M.; Sharpe, M.L.; Francisco, L.M.; Sharpe, A.H.; Blazar, B.R.; Munn, D.H.; et al. Physiologic Control of IDO Competence in Splenic Dendritic Cells. J. Immunol. 2011, 187, 2329–2335. [Google Scholar] [CrossRef] [Green Version]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic Phenotype of IFN-gamma-Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.; Longman, R.S.; Albert, M.L. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood 2005, 106, 2375–2381. [Google Scholar] [CrossRef] [Green Version]
- Jung, I.D.; Lee, J.S.; Lee, C.M.; Noh, K.T.; Jeong, Y.I.; Park, W.S.; Chun, S.H.; Jeong, S.K.; Park, J.W.; Son, K.H.; et al. Induction of indoleamine 2,3-dioxygenase expression via heme oxygenase-1-dependant pathway during murine dendritic cell maturation. Biochem. Pharmacol. 2010, 80, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Bracho-Sanchez, E.; Hassanzadeh, A.; Brusko, M.A.; Wallet, M.A.; Keselowsky, B.G. Dendritic Cells Treated with Exogenous Indoleamine 2,3-Dioxygenase Maintain an Immature Phenotype and Suppress Antigen-specific T cell Proliferation. J. Immunol. Regen. Med. 2019, 5, 100015. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Gao, F.; Gu, K.; Chen, D.K. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2019, 10, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, S.K.; Egilmez, N.G.; Suttles, J.; Stout, R.D. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophagesin vitro and in vivo. J. Immunol. 2007, 178, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Soldano, S.P.; Trombetta, C.P.; Contini, P.P.; Tomatis, V.M.; Ruaro, B.M.; Brizzolara, R.P.; Montagna, P.; Sulli, A.; Paolino, S.; Pizzorni, C.; et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1842–1845. [Google Scholar] [CrossRef]
- Jiang, N.; Zhang, L.; Zhao, G.; Lin, J.; Wang, Q.; Xu, Q.; Li, C.; Hu, L.; Peng, X.; Yu, F.; et al. Indoleamine 2,3-Dioxygenase Regulates Macrophage Recruitment, Polarization and Phagocytosis in Aspergillus Fumigatus Keratitis. Investig. Ophthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Naka, T.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 2009, 206, 2027–2035. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.S.; Adu-Gyamfi, C.G.; Cumming, B.M.; Savulescu, D.M. Evolutionary Views of Tuberculosis: Indoleamine 2,3-Dioxygenase Catalyzed Nicotinamide Synthesis Reflects Shifts in Macrophage Metabolism: Indoleamine 2,3-Dioxygenase Reflects Altered Macrophage Metabolism During Tuberculosis Pathogenesis. Bioessays 2020, 42, e1900220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.S.; Passey, R.; Matanovic, G.; Smythe, G.; Kapoor, V. Evidence for increased de novo synthesis of NAD in immune-activated RAW264.7 macrophages: A self-protective mechanism? Arch. Biochem. Biophys. 1999, 372, 1–7. [Google Scholar] [CrossRef]
- Mandal, A.; Viswanathan, C. Natural killer cells: In health and disease. Hematol. Oncol. Stem Cell Ther. 2015, 8, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Shah, M.H.; Turner, M.J.; VanDeusen, J.B.; Whitman, S.P.; Cooper, M.A.; Suzuki, K.; Wechser, M.; Goodsaid, F.; Caligiuri, M.A. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: Implications for the innate immune response. J. Immunol. 1999, 162, 4511–4520. [Google Scholar]
- Arase, N.; Arase, H.; Hirano, S.; Yokosuka, T.; Sakurai, D.; Saito, T. IgE-mediated activation of NK cells through Fc gamma RIII. J. Immunol. 2003, 170, 3054–3058. [Google Scholar] [CrossRef] [Green Version]
- Tirado-Gonzalez, I.; Barrientos, G.; Freitag, N.; Otto, T.; Thijssen, V.L.; Moschansky, P.; Von Kwiatkowski, P.; Klapp, B.F.; Winterhager, E.; Bauersachs, S.; et al. Uterine NK cells are critical in shaping DC immunogenic functions compatible with pregnancy progression. PLoS ONE 2012, 7, e46755. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, T.E.; Sun, J.C.; Lanier, L.L. Natural killer cell memory. Immunity 2015, 43, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imada, K.; Leonard, W.J. The Jak-STAT pathway. Mol. Immunol. 2000, 37, 1–11. [Google Scholar] [CrossRef]
- Gotthardt, D.; Sexl, V. STATs in NK-cells: The good, the bad, and the ugly. Front. Immunol. 2016, 7, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masselli, E.; Vaccarezza, M.; Carubbi, C.; Pozzi, G.; Presta, V.; Mirandola, P.; Vitale, M. NK cells: A double edge sword against SARS-CoV-2. Adv. Biol. Regul. 2020, 77, 100737. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liang, S.; Zhang, C. NK Cells in Autoimmune Diseases: Protective or Pathogenic? Front. Immunol. 2021, 12, 624687. [Google Scholar] [CrossRef]
- Kai, S.; Goto, S.; Tahara, K.; Sasaki, A.; Tone, S.; Kitano, S. Indoleamine 2,3-dioxygenase is necessary for cytolytic activity of natural killer cells. Scand. J. Immunol. 2004, 59, 177–182. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Yang, Y.; Lee, Y.; Kim, M.S.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Indoleamine-2,3-Dioxygenase in Thyroid Cancer Cells Suppresses Natural Killer Cell Function by Inhibiting NKG2D and NKp46 Expression via STAT Signaling Pathways. J. Clin. Med. 2019, 8, 842. [Google Scholar] [CrossRef] [Green Version]
- Hogan, S.P. Functional role of eosinophils in gastrointestinal inflammation. Immunol. Allergy Clin. N. Am. 2009, 29, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef]
- Wen, T.; Rothenberg, M.E. The Regulatory Function of Eosinophils. Microbiol. Spectr. 2016, 4, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.J.; Woo, S.Y.; Jang, M.H.; Miyasaka, M.; Ryu, K.H.; Park, H.K.; Seoh, J.Y. Human eosinophils show chemotaxis to lymphoid chemokines and exhibit antigen-presenting-cell-like properties upon stimulation with IFN-gamma, IL-3 and GM-CSF. Int. Arch. Allergy Immunol. 2008, 146, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, R.; Lotze, M.T. Eosinophils induce DC maturation, regulating immunity. J. Leukoc. Biol. 2008, 83, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.Z. Eosinophils function as antygen presenting cells. J. Leukoc. Biol. 2004, 76, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Venge, P. The eosinophil and airway remodelling in asthma. Clin. Respir. J. 2010, 4 (Suppl. S1), 15–19. [Google Scholar] [CrossRef]
- Astigiano, S.; Morandi, B.; Costa, R.; Mastracci, L.; D’Agostin, A.; Ratto, G.B.; Melioli, G.; Frumento, G. Eosinophil granulocytes account for indoleamine 2,3-dioxygenase-mediated immune escape in human non-small cell lung cancer. Neoplasia 2005, 7, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odemuyiwa, S.O.; Ebeling, C.; Duta, V.; Abel, M.; Puttagunta, L.; Cravetchi, O.; Majaesic, C.; Vliagoftis, H.; Moqbel, R. Tryptophan catabolites regulate mucosal sensitization to ovalbumin in respiratory airways. Allergy 2009, 64, 488–492. [Google Scholar] [CrossRef]
- Spencer, L.A.; Szela, C.T.; Perez, S.A.; Kirchhoffer, C.L.; Neves, J.S.; Radke, A.L.; Weller, P.F. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J. Leukoc. Biol. 2009, 85, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Tulic, M.K.; Sly, P.D.; Andrews, D.; Crook, M.; Davoine, F.; Odemuyiwa, S.O.; Charles, A.; Hodder, M.L.; Prescott, S.L.; Holt, P.G.; et al. Thymic indoleamine 2,3-dioxygenase-positive eosinophils in young children: Potential role in maturation of the naive immune system. Am. J. Pathol. 2009, 175, 2043–2052. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Sheppard, F.R.; Kelher, M.R.; Moore, E.E.; McLaughlin, N.J.; Banerjee, A.; Silliman, C.C. Structural organization of the neutrophil NADPH oxidase: Phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 2005, 78, 1025–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef]
- Kim, M.H.; Liu, W.; Borjesson, D.L.; Curry, F.R.; Miller, L.S.; Cheung, A.L.; Liu, F.T.; Isseroff, R.R.; Simon, S.I. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J. Investig. Dermatol. 2008, 128, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scapini, P.; Bazzoni, F.; Cassatella, M.A. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol. Lett. 2008, 116, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pillay, J.; Kamp, V.M.; Van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Abi Abdallah, D.S.; Egan, C.E.; Butcher, B.A.; Denkers, E.Y. Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int. Immunol. 2011, 23, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauvillain, C.; Delneste, Y.; Scotet, M.; Peres, A.; Gascan, H.; Guermonprez, P.; Barnaba, V.; Jeannin, P. Neutrophils efficiently cross-prime naive T cells in vivo. Blood 2007, 110, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; Von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defense, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novitskiy, S.V.; Pickup, M.W.; Gorska, A.E.; Owens, P.; Chytil, A.; Aakre, M.; Wu, H.; Shyr, Y.; Moses, H.L. TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov. 2011, 1, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughman, J.A.; Hunstad, D.A. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microbes Infect. 2011, 13, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughman, J.A.; Hunstad, D.A. Induction of indoleamine 2,3- dioxygenase by uropathogenic bacteria attenuates innate responses to epithelial infection. J. Infect. Dis. 2012, 205, 1830–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo, A.F.; Read, R.C.; Arevalo-Herrera, M.; Herrera, S.; Elliott, T.; Polak, M.E. Malaria systems immunology: Plasmodium vivax induces tolerance during primary infection through dysregulation of neutrophils and dendritic cells. J. Infect. 2018, 77, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Lewkowicz, N.; Klink, M.; Mycko, M.P.; Lewkowicz, P. Neutrophil--CD4+CD25+ T regulatory cell interactions: A possible new mechanism of infectious tolerance. Immunobiology 2013, 218, 455–464. [Google Scholar] [CrossRef]
- Babcock, T.A.; Carlin, J.M. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine 2000, 12, 588–594. [Google Scholar] [CrossRef]
- Johansson, A.S.; Owe-Larsson, B.; Asp, L.; Kocki, T.; Adler, M.; Hetta, J.; Gardner, R.; Lundkvist, G.B.; Urbanska, E.M.; Karlsson, H. Activation of kynurenine pathway in ex vivo fibroblasts from patients with bipolar disorder or schizophrenia: Cytokine challenge increases production of 3-hydroxykynurenine. J. Psychiatr. Res. 2013, 47, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar]
- Nino-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immun. 2014, 20, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Hettler, V.; Fischer, V.; Mawrin, C.; Hartig, R.; Dobrowolny, H.; Bogerts, B.; Frodl, T.; Busse, S. Increased quinolinic acid in peripheral mononuclear cells in Alzheimer’s dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 268, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Rossez, H.; Lim, C.K.; Jugder, B.E.; Brew, B.J.; Guillemin, G.J. Characterization of the Kynurenine Pathway in CD8(+) Human Primary Monocyte-Derived Dendritic Cells. Neurotox. Res. 2016, 30, 620–632. [Google Scholar] [CrossRef]
- McIlroy, D.; Tanguy-Royer, S.; Le Meur, N.; Guisle, I.; Royer, P.J.; Leger, J.; Meflah, K.; Gregoire, M. Profiling dendritic cell maturation with dedicated microarrays. J. Leukoc. Biol. 2005, 78, 794–803. [Google Scholar] [CrossRef]
- Loughman, J.A.; Yarbrough, M.L.; Tiemann, K.M.; Hunstad, D.A. Local Generation of Kynurenines Mediates Inhibition of Neutrophil Chemotaxis by Uropathogenic Escherichia coli. Infect. Immun. 2016, 84, 1176–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, T.; Saito, K.; Takemura, M.; Maekawa, N.; Fujigaki, S.; Fujii, H.; Wada, H.; Takeuchi, S.; Noma, A.; Seishima, M. 3-Hydroxyanthranilic acid, an L-tryptophan metabolite, induces apoptosis in monocyte-derived cells stimulated by interferon-gamma. Ann. Clin. Biochem. 2001, 38, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ye, Z.; Kijlstra, A.; Zhou, Y.; Yang, P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2014, 177, 521–530. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Romagnani, S. T-cell subsets (Th1 versus Th2). Ann. Allergy Asthma Immunol. 2000, 85, 9–18. [Google Scholar] [CrossRef]
- Illera, V.A.; Perandones, C.E.; Stunz, L.L.; Mower, D.A., Jr.; Ashman, R.F. Apoptosis in splenic B lymphocytes. Regulation by protein kinase C and IL-4. J. Immunol. 1993, 151, 2965–2973. [Google Scholar]
- Gu, C.; Wu, L.; Li, X. IL-17 family: Cytokines, receptors and signaling. Cytokine 2013, 64, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Korn, T.; Oukka, M.; Kuchroo, V.K. Induction and effector functions of TH17 cells. Nature 2008, 453, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, G.; Miossec, P. Interleukin 17 contributes to the chronicity of inflammatory diseases such as rheumatoid arthritis. Eur. J. Immunol. 2014, 44, 339–347. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Guo, B. IL-10 Modulates Th17 Pathogenicity during Autoimmune Diseases. J. Clin. Cell. Immunol. 2016, 7, 400. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 2015, 69, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Nguyen, L.P.; Noh, S.K.; Bray, T.M.; Bruno, R.S.; Ho, E. Induction of regulatory T cells by green tea polyphenol EGCG. Immunol. Lett. 2011, 139, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Jonuleit, H.; Schmitt, E. The regulatory T cell family: Distinct subsets and their interrelations. J. Immunol. 2003, 171, 6323–6327. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.Y.; Flavell, R.A. TGF-beta and regulatory T cell in immunity and autoimmunity. J. Clin. Immunol. 2008, 28, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Pot, C.; Apetoh, L.; Kuchroo, V.K. Type 1 regulatory T cells (Tr1) in autoimmunity. Semin. Immunol. 2011, 23, 202–208. [Google Scholar] [CrossRef] [Green Version]
- De Waal Malefyt, R.; Haanen, J.; Spits, H.; Roncarolo, M.G.; Te Velde, A.; Figdor, C.; Johnson, K.; Kastelein, R.; Yssel, H.; De Vries, J.E. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 1991, 174, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Yuan, J.; Kuchroo, V.K.; Weiner, H.L. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. J. Immunol. 2007, 178, 179–185. [Google Scholar] [CrossRef]
- Chen, M.L.; Yan, B.S.; Bando, Y.; Kuchroo, V.K.; Weiner, H.L. Latency-associated peptide identifies a novel CD4+CD25+ regulatory T cell subset with TGFbeta-mediated function and enhanced suppression of experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 7327–7337. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. T(reg) cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Uyttenhove, C.; Van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Filippi, C.M.; Juedes, A.E.; Oldham, J.E.; Ling, E.; Togher, L.; Peng, Y.; Flavell, R.A.; Von Herrath, M.G. Transforming growth factor-beta suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: A two-faced impact on autoimmunity. Diabetes 2008, 57, 2684–2692. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Mellor, A.L. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol. 2004, 172, 4100–4110. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Indoleamine 2,3-dioxygenase depletes tryptophan, activates general control non-derepressible 2 kinase and down-regulates key enzymes involved in fatty acid synthesis in primary human CD4+ T cells. Immunology 2015, 146, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 eIF2kinaseis required for adaptation to amino acid deprivation in mice. Mol. Cell. Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Tsogka, K.; Sounidaki, M.; Liakopoulos, V.; Stefanidis, I. Indoleamine 2,3-dioxygenase downregulates T-cell receptor complex zeta-chain and c-Myc, and reduces proliferation, lactate dehydrogenase levels and mitochondrial glutaminase in human T-cells. Mol. Med. Rep. 2016, 13, 925–932. [Google Scholar] [CrossRef]
- Hayashi, T.; Mo, J.H.; Gong, X.; Rossetto, C.; Jang, A.; Beck, L.; Elliott, G.I.; Kufareva, I.; Abagyan, R.; Broide, D.H.; et al. 3-hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 18619–18624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Sounidaki, M.; Tsogka, K.; Antoniadis, N.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Indoleamine2,3-dioxygenase, by degrading L-tryptophan, enhances carnitine palmitoyltransferase I activity and fatty acid oxidation, and exerts fatty acid-dependent effects in human allo-reactive CD4+ T-cells. Int. J. Mol. Med. 2016, 38, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. IDO decreases glycolysis and glutaminolysis by activating GCN2K, while it increases fatty acid oxidation by activating AhR, thus preserving CD4+ T-cell survival and proliferation. Int. J. Mol. Med. 2018, 42, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Molano, A.; Illarionov, P.A.; Besra, G.S.; Putterman, C.; Porcelli, S.A. Modulation of invariant natural killer T cell cytokine responses by indoleamine 2,3-dioxygenase. Immunol. Lett. 2008, 117, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refaeli, Y.; Van Parijs, L.; Alexander, S.I.; Abbas, A.K. Interferon gamma is required for activation-induced death of T lymphocytes. J. Exp. Med. 2002, 196, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Oriss, T.B.; Fei, M.; Henry, A.C.; Melgert, B.N.; Chen, L.; Mellor, A.L.; Munn, D.H.; Irvin, C.G.; Ray, P.; et al. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 6690–6695. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Beck, L.; Rossetto, C.; Gong, X.; Takikawa, O.; Takabayashi, K.; Broide, D.H.; Carson, D.A.; Raz, E. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J. Clin. Investig. 2004, 114, 270–279. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, C.R.; Heseler, K.; Muller, A.; Daubener, W. Role of indoleamine 2,3-dioxygenase in antimicrobial defence and immuno-regulation: Tryptophan depletion versus production of toxic kynurenines. Curr. Drug Metab. 2007, 8, 237–244. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 2003, 527, 183–190. [Google Scholar] [PubMed]
- Lee, S.M.; Lee, Y.S.; Choi, J.H.; Park, S.G.; Choi, I.W.; Joo, Y.D.; Lee, W.S.; Lee, J.N.; Choi, I.; Seo, S.K. Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cel death via intracellular GSH depletion. Immunol. Lett. 2010, 132, 53–60. [Google Scholar] [CrossRef]
- Orihara, K.; Odemuyiwa, S.O.; Stefura, W.P.; Ilarraza, R.; HayGlass, K.T.; Moqbel, R. Neurotransmitter signalling via NMDA receptors leads to decreased T helper type 1-like and enhanced T helper type 2-like immune balance in humans. Immunology 2018, 153, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Negishi, T.; Kato, Y.; Ooneda, O.; Mimura, J.; Takada, T.; Mochizuki, H.; Yamamoto, M.; Fujii-Kuriyama, Y.; Furusako, S. Effects of aryl hydrocarbon receptor signaling on the modulation of TH1/TH2 balance. J. Immunol. 2005, 175, 7348–7356. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Ambrosio, L.F.; Insfran, C.; Volpini, X.; Acosta Rodriguez, E.; Serra, H.M.; Quintana, F.J.; Cervi, L.; Motrán, C.C. Role of Aryl Hydrocarbon Receptor (AhR) in the Regulation of Immunity and Immunopathology During Trypanosoma cruzi Infection. Front. Immunol. 2019, 10, 631. [Google Scholar] [CrossRef] [Green Version]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [Green Version]
- Lane, N.; Robins, R.A.; Corne, J.; Fairclough, L. Regulation in chronic obstructive pulmonary disease: The role of regulatory T-cells and T17 cells. Clin. Sci. (Lond.) 2010, 119, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Hu, G.H.; Kang, H.Y.; Yao, H.B.; Kou, W.; Liu, H.; Zhang, C.; Hong, S.L. An aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress the Th17 response in allergic rhinitis patients. Lab. Investig. 2014, 94, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.G.; Wu, X.C.; Yang, J.S.; Xu, L.Y.; Liu, X.; Huang, Y.M.; Bjelke, B.; Link, H. Therapeutic potential of IFN-gamma-modified dendritic cells in acute and chronic experimental allergic encephalomyelitis. Int. Immunol. 2004, 16, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.G.; Liu, X.; Link, H. Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids 2004, 69, 653–659. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, G.X.; Gran, B.; Fallarino, F.; Yu, S.; Li, H.; Cullimore, M.L.; Rostami, A.; Xu, H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 5953–5961. [Google Scholar] [CrossRef]
- Salimi Elizei, S.; Poormasjedi-Meibod, M.S.; Wang, X.; Kheirandish, M.; Ghahary, A. Kynurenic acid downregulates IL-17/IL-23 axis in vitro. Mol. Cell. Biochem. 2017, 431, 55–65. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, E.D.; Engin, A. The effect of environmental pollution on immune evasion checkpoints of SARS-CoV-2. Environ. Toxicol. Pharmacol. 2021, 81, 103520. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Hou, D.Y.; Liu, Y.; Koni, P.A.; Metz, R.; Chandler, P.; Mellor, A.L.; He, Y.; Munn, D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009, 113, 6102–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platten, M.; Ho, P.P.; Youssef, S.; Fontoura, P.; Garren, H.; Hur, E.M.; Gupta, R.; Lee, L.Y.; Kidd, B.A.; Robinson, W.H.; et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 2005, 310, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazio, F.; Zappulla, C.; Notartomaso, S.; Busceti, C.; Bessede, A.; Scarselli, P.; Vacca, C.; Gargaro, M.; Volpi, C.; Allegrucci, M.; et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology 2014, 81, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Lefrancois, L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003, 3, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Dagenais-Lussier, X.; Aounallah, M.; Mehraj, V.; El-Far, M.; Tremblay, C.; Sekaly, R.P.; Routy, J.P.; Van Grevenynghe, J. Kynurenine Reduces Memory CD4 T-Cell Survival by Interfering with Interleukin-2 Signaling Early during HIV-1 Infection. J. Virol. 2016, 90, 7967–7979. [Google Scholar] [CrossRef] [Green Version]
- Belladonna, M.L.; Grohmann, U.; Guidetti, P.; Volpi, C.; Bianchi, R.; Fioretti, M.C.; Schwarcz, R.; Fallarino, F.; Puccetti, P. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J. Immunol. 2006, 177, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, R.B.; Berge-Hansen, L.; Junker, N.; Hansen, C.A.; Hadrup, S.R.; Schumacher, T.N.; Svane, I.M.; Becker, J.C.; Thor Straten, P.; Andersen, M.H. The immune system strikes back: Cellular immune responses against indoleamine 2,3-dioxygenase. PLoS ONE 2009, 4, e6910. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, R.B.; Hadrup, S.R.; Svane, I.M.; Hjortso, M.C.; Thor Straten, P.; Andersen, M.H. Indoleamine 2,3-dioxygenase specific, cytotoxic T cells as immune regulators. Blood 2011, 117, 2200–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, S.; Larsen, S.K.; Iversen, T.Z.; Donia, M.; Klausen, T.W.; Svane, I.M.; Thor Straten, P.; Andersen, M.H. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS ONE 2012, 7, e34568. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, J.D.; Le Buanec, H.; Saussine, A.; Bensussan, A.; Bagot, M. IL-10 producing regulatory B cells in mice and humans: State of the art. Curr. Mol. Med. 2012, 12, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, S.; Morva, A.; Youinou, P.; Jamin, C. Regulatory B cells in autoimmune diseases: How do they work? Ann. N. Y. Acad. Sci. 2009, 1173, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Chesneau, M.; Michel, L.; Degauque, N.; Brouard, S. Regulatory B cells and tolerance in transplantation: From animal models to human. Front. Immunol. 2013, 4, 497. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.N.; DuHadaway, J.; Pigott, E.; Ridge, N.; Prendergast, G.C.; Muller, A.J.; Mandik-Nayak, L. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J. Immunol. 2009, 182, 7509–7517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, D.S.; Kim, C.H.; Chang, K.H.; Kwon, B.S. PDCA expression by B lymphocytes reveals important functional attributes. J. Immunol. 2010, 184, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.G.; Wang, J.H.; Stohl, W.; Kim, K.S.; Gray, J.D.; Horwitz, D.A. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J. Immunol. 2006, 176, 3321–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin-Ethier, J.; Hanafi, L.A.; Duvignaud, J.B.; Leclerc, D.; Lapointe, R. IDO expression by human B lymphocytes in response to T lymphocyte stimuli and TLR engagement is biologically inactive. Mol. Immunol. 2011, 49, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.; Dahlke, K.; Sontheimer, K.; Hagn, M.; Kaltenmeier, C.; Barth, T.F.; Beyer, T.; Reister, F.; Fabricius, D.; Lotfi, R.; et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013, 73, 2468–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Chen, X.; Liu, Q.; Zhang, X.; Huang, K.; Liu, L.; Li, H.; Zhou, M.; Huang, F.; Fan, Z.; et al. Mesenchymal stromal cells infusions improve refractory chronic graft versus host disease through an increase of CD5+ regulatory B cells producing interleukin 10. Leukemia 2015, 29, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Piper, C.J.M.; Rosser, E.C.; Oleinika, K.; Nistala, K.; Krausgruber, T.; Rendeiro, A.F.; Banos, A.; Drozdov, I.; Villa, M.; Thomson, S.; et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep. 2019, 29, 1878–1892.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, L.M.; Pigott, E.; Duhadaway, J.B.; Grabler, S.; Metz, R.; Prendergast, G.C.; Mandik-Nayak, L. IDO2 is a critical mediator of autoantibody production and inflammatory pathogenesis in a mouse model of autoimmune arthritis. J. Immunol. 2014, 192, 2082–2090. [Google Scholar] [CrossRef] [Green Version]
- Merlo, L.M.; Grabler, S.; DuHadaway, J.B.; Pigott, E.; Manley, K.; Prendergast, G.C.; Laury-Kleintop, L.D.; Mandik-Nayak, L. Therapeutic antibody targeting of indoleamine-2,3-dioxygenase (IDO2) inhibits autoimmune arthritis. Clin. Immunol. 2017, 179, 8–16. [Google Scholar] [CrossRef]
- Merlo, L.M.; Bowers, J.; Stefanoni, T.; Getts, R.; Mandik-Nayak, L. B-Cell-Targeted 3DNA Nanotherapy Against Indoleamine 2,3-Dioxygenase 2 (IDO2) Ameliorates Autoimmune Arthritis in a Preclinical Model. Clin. Pathol. 2020, 13, 2632010X20951812. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; DuHadaway, J.B.; Montgomery, J.D.; Peng, W.D.; Murray, P.J.; Prendergast, G.C.; Caton, A.J.; Muller, A.J.; Mandik-Nayak, L. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Front. Immunol. 2020, 11, 1861. [Google Scholar] [CrossRef]
- Hober, D.; Sauter, P. Pathogenesis of type 1 diabetes mellitus: Interplay between enterovirus and host. Nat. Rev. Endocrinol. 2010, 6, 279–289. [Google Scholar] [CrossRef]
- Eisenbarth, G.S. Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 1986, 314, 1360–1368. [Google Scholar]
- Bonifacio, E. Predicting type 1 diabetes using biomarkers. Diabetes Care 2015, 38, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef]
- Virostko, J.; Williams, J.; Hilmes, M.; Bowman, C.; Wright, J.J.; Du, L.; Kang, H.; Russell, W.E.; Powers, A.C.; Moore, D.J. Pancreas volume declines during the first year after diagnosis of type 1 diabetes and exhibits altered diffusion at disease onset. Diabetes Care 2019, 42, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerovich, K.; Ortis, F.; Allagnat, F.; Cardozo, A.K. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J. Mol. Endocrinol. 2016, 57, R1–R17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roep, B.O.; Thomaidou, S.; Van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system)? Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef]
- Culina, S.; Lalanne, A.I.; Afonso, G.; Cerosaletti, K.; Pinto, S.; Sebastiani, G.; Kuranda, K.; Nigi, L.; Eugster, A.; Østerbye, T.; et al. Islet-reactive CD8+ T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Tree, T.I.; Lawson, J.; Edwards, H.; Skowera, A.; Arif, S.; Roep, B.O.; Peakman, M. Naturally arising human CD4 T-cells that recognize islet autoantigens and secrete interleukin-10 regulate proinflammatory T-cell responses via linked suppression. Diabetes 2010, 59, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- De Filette, J.M.K.; Pen, J.J.; Decoster, L.; Vissers, T.; Bravenboer, B.; Van Der Auwera, B.J.; Gorus, F.K.; Roep, B.O.; Aspeslagh, S.; Neyns, B.; et al. Immune checkpoint inhibitors and type 1 diabetes mellitus: A case report and systematic review. Eur. J. Endocrinol. 2019, 181, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, F.; Yue, T.T.; Wang, F.X.; Yang, C.L.; Luo, J.H.; Rong, S.J.; Xiong, F.; Zhang, S.; Wang, C.Y. Revisiting the Antigen-Presenting function of β Cells in T1D Pathogenesis. Front. Immunol. 2021, 12, 690783. [Google Scholar] [CrossRef]
- Peters, L.; Posgai, A.; Brusko, T.M. Islet-immune interactions in type 1 diabetes: The nexus of beta cell destruction. Clin. Exp. Immunol. 2019, 198, 326–340. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.W.; Terentis, A.C.; King, N.J.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef] [PubMed]
- Poormasjedi-Meibod, M.S.; Jalili, R.B.; Hosseini-Tabatabaei, A.; Hartwell, R.; Ghahary, A. Immuno-regulatory function of indoleamine 2,3 dioxygenase through modulation of innate immune responses. PLoS ONE 2013, 8, e71044. [Google Scholar]
- Atkinson, M.A.; Leiter, E.H. The NOD mouse model of type 1 diabetes: As good as it gets? Nat. Med. 1999, 5, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, H.; Sprent, J. A defect in central tolerance in NOD mice. Nat. Immunol. 2001, 2, 1025–1031. [Google Scholar] [CrossRef]
- Serreze, D.V.; Gaedeke, J.W.; Leiter, E.H. Hematopoietic stem-cell defects underlying abnormal macrophage development and maturation in NOD/Lt mice: Defective regulation of cytokine receptors and protein kinase C. Proc. Natl. Acad. Sci. USA 1993, 90, 9625–9629. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Wu, D.; Swinney, C.; Elliott, R.B. Immunohistochemical analyses of pancreatic macrophages and CD4 and CD8 T cell subsets prior to and following diabetes in the NOD mouse. Pancreas 1995, 11, 16–25. [Google Scholar] [CrossRef]
- Puccetti, P.; Grohmann, U. IDO and regulatory T cells: A role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 2007, 7, 817–823. [Google Scholar] [CrossRef]
- Nikolic, T.; Welzen-Coppens, J.M.; Leenen, P.J.; Drexhage, H.A.; Versnel, M.A. Plasmacytoid dendritic cells in autoimmune diabetes—Potential tools for immunotherapy. Immunobiology 2009, 214, 791–799. [Google Scholar] [CrossRef]
- Grohmann, U.; Fallarino, F.; Bianchi, R.; Vacca, C.; Orabona, C.; Belladonna, M.L.; Fioretti, M.C.; Puccetti, P. Tryptophan catabolism in nonobese diabetic mice. Adv. Exp. Med. Biol. 2003, 527, 47–54. [Google Scholar]
- Grohmann, U.; Fallarino, F.; Bianchi, R.; Orabona, C.; Vacca, C.; Fioretti, M.C.; Puccetti, P. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J. Exp. Med. 2003, 198, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Bianchi, R.; Orabona, C.; Vacca, C.; Belladonna, M.L.; Fioretti, M.C.; Serreze, D.V.; Grohmann, U.; Puccetti, P. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J. Exp. Med. 2004, 200, 1051–1062. [Google Scholar] [CrossRef] [Green Version]
- Hosseini-Tabatabaei, A.; Jalili, R.B.; Li, Y.; Kilani, R.T.; Moeen Rezakhanlou, A.; Ghahary, A. Mechanism underlying defective interferon gamma-induced IDO expression in non-obese diabetic mouse fibroblasts. PLoS ONE 2012, 7, e37747. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Volpi, C.; Zelante, T.; Vacca, C.; Calvitti, M.; Fioretti, M.C.; Puccetti, P.; Romani, L.; Grohmann, U. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J. Immunol. 2009, 183, 6303–6312. [Google Scholar] [CrossRef] [Green Version]
- Pallotta, M.T.; Orabona, C.; Bianchi, R.; Vacca, C.; Fallarino, F.; Belladonna, M.L.; Volpi, C.; Mondanelli, G.; Gargaro, M.; Allegrucci, M.; et al. Forced IDO1 expression in dendritic cells restores immunoregulatory signalling in autoimmune diabetes. J. Cell Mol. Med. 2014, 18, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Albini, E.; Pallotta, M.T.; Volpi, C.; Chatenoud, L.; Kuhn, C.; Fallarino, F.; Matino, D.; Belladonna, M.L.; Bianchi, R.; et al. The proteasome inhibitor Bortezomib controls Indoleamine 2,3-dioxygenase 1 breakdown and restores immune regulation in autoimmune diabetes. Front. Immunol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jalili, R.B.; Kilani, R.T.; Elizei, S.S.; Farrokhi, A.; Khosravi-Maharlooei, M.; Warnock, G.L.; Ao, Z.; Marzban, L.; Ghahary, A. IDO-Expressing Fibroblasts Protect Islet Beta Cells From Immunological Attack and Reverse Hyperglycemia in Non-Obese Diabetic Mice. J. Cell Physiol. 2016, 231, 1964–1973. [Google Scholar] [CrossRef]
- Alexander, A.M.; Crawford, M.; Bertera, S.; Rudert, W.A.; Takikawa, O.; Robbins, P.D.; Trucco, M. Indoleamine 2,3-dioxygenase expression in transplanted NOD islets prolongs graft survival after adoptive transfer of diabetogenic splenocytes. Diabetes 2002, 51, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Bertera, S.; Alexander, A.M.; Crawford, M.L.; Papworth, G.; Watkins, S.C.; Robbins, P.D.; Trucco, M. Gene combination transfer to block autoimmune damage in transplanted islets of Langerhans. Exp. Diabesity Res. 2004, 5, 201–210. [Google Scholar] [CrossRef]
- Fallarino, F.; Luca, G.; Calvitti, M.; Mancuso, F.; Nastruzzi, C.; Fioretti, M.C.; Grohmann, U.; Becchetti, E.; Burgevin, A.; Kratzer, R.; et al. Therapy of experimental type 1 diabetes by isolated Sertoli cell xenografts alone. J. Exp. Med. 2009, 206, 2511–2526. [Google Scholar] [CrossRef] [Green Version]
- Ueno, A.; Cho, S.; Cheng, L.; Wang, J.; Hou, S.; Nakano, H.; Santamaria, P.; Yang, Y. Transient Upregulation of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Human Chorionic Gonadotropin Downregulates Autoimmune Diabetes. Diabetes 2007, 56, 1686–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, H.; Mohamed, E.; Huang, L.; Chandler, P.R.; Ou, R.; Pacholczyk, R.; Mellor, A.L. Stimulator of interferon genes agonists attenuate type I diabetes progression in NOD mice. Immunology 2019, 158, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Sun, F.; Yang, C.; Wang, F.; Luo, J.; Yang, P.; Xiong, F.; Zhang, S.; Yu, Q.; Wang, C.Y. The AHR Signaling Attenuates Autoimmune Responses During the Development of Type 1 Diabetes. Front. Immunol. 2020, 11, 1510. [Google Scholar] [CrossRef] [PubMed]
- Kerkvliet, N.I.; Steppan, L.B.; Vorachek, W.; Oda, S.; Farrer, D.; Wong, C.P.; Pham, D.; Mourich, D.V. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy 2009, 1, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Bergerot, I.; Ploix, C.; Petersen, J.; Moulin, V.; Rask, C.; Fabien, N.; Lindblad, M.; Mayer, A.; Czerkinsky, C.; Holmgren, J.; et al. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1997, 94, 4610–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspord, C.; Thivolet, C. Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 2002, 130, 204–211. [Google Scholar] [CrossRef]
- Ploix, C.; Bergerot, I.; Durand, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes 1999, 48, 2150–2156. [Google Scholar] [CrossRef]
- Kim, N.S.; Mbongue, J.C.; Nicholas, D.A.; Esebanmen, G.E.; Unternaehrer, J.J.; Firek, A.F.; Langridge, W.H. Chimeric Vaccine Stimulation of Human Dendritic Cell Indoleamine 2, 3-Dioxygenase Occurs via the Non-Canonical NF-κB Pathway. PLoS ONE 2016, 11, e0147509. [Google Scholar] [CrossRef] [Green Version]
- Ghazarian, L.; Diana, J.; Beaudoin, L.; Larsson, P.G.; Puri, R.K.; Van Rooijen, N.; Flodström-Tullberg, M.; Lehuen, A. Protection against type 1 diabetes upon Coxsackievirus B4 infection and iNKT-cell stimulation: Role of suppressive macrophages. Diabetes 2013, 62, 3785–3796. [Google Scholar] [CrossRef] [Green Version]
- Dolpady, J.; Sorini, C.; Di Pietro, C.; Cosorich, I.; Ferrarese, R.; Saita, D.; Clementi, M.; Canducci, F.; Falcone, M. Oral Probiotic VSL#3 Prevents Autoimmune Diabetes by Modulating Microbiota and Promoting Indoleamine 2,3-Dioxygenase-Enriched Tolerogenic Intestinal Environment. J. Diabetes Res. 2016, 2016, 7569431. [Google Scholar]
- Soichot, M.; Hennart, B.; Al Saabi, A.; Leloire, A.; Froguel, P.; Levy-Marchal, C.; Poulain-Godefroy, O.; Allorge, D. Identification of a variable number of tandem repeats polymorphism and characterization of LEF-1 response elements in the promoter of the IDO1 gene. PLoS ONE 2011, 6, e25470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.K.; Simon, J.S.; Gustafson, E.L.; Noviello, S.; Cubells, J.F.; Epstein, M.P.; Devlin, D.J.; Qiu, P.; Albrecht, J.K.; Brass, C.A.; et al. Association of a polymorphism in the indoleamine- 2,3-dioxygenase gene and interferon-α-induced depression in patients with chronic hepatitis C. Mol. Psychiatry 2012, 17, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Orabona, C.; Mondanelli, G.; Pallotta, M.T.; Carvalho, A.; Albini, E.; Fallarino, F.; Vacca, C.; Volpi, C.; Belladonna, M.L.; Berioli, M.G.; et al. Deficiency of immunoregulatory indoleamine 2,3-dioxygenase 1 in juvenile diabetes. JCI Insight 2018, 3, e96244. [Google Scholar] [CrossRef] [PubMed]
- Anquetil, F.; Mondanelli, G.; Gonzalez, N.; Rodriguez Calvo, T.; Zapardiel Gonzalo, J.; Krogvold, L.; Dahl-Jørgensen, K.; Van Den Eynde, B.; Orabona, C.; Grohmann, U.; et al. Loss of IDO1 expression from human pancreatic beta-cells precedes their destruction during the development of type 1 diabetes. Diabetes 2018, 67, 1858–1866. [Google Scholar] [CrossRef] [Green Version]
- Zoso, A.; Mazza, E.M.; Bicciato, S.; Mandruzzato, S.; Bronte, V.; Serafini, P.; Inverardi, L. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur. J. Immunol. 2014, 44, 3307–3319. [Google Scholar] [CrossRef]
- Badal, D.; Dayal, D.; Singh, G.; Sachdeva, N. Role of DNA-LL37 complexes in the activation of plasmacytoid dendritic cells and monocytes in subjects with type 1 diabetes. Sci. Rep. 2020, 10, 8896. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mao, C.; Zhao, Z.; Gu, Q.; Jin, M.; Xiao, Y.; Jiang, X.; Zhao, Y.; Zhang, Y.; Ning, G. Increased TTS abrogates IDO-mediated CD4+ T cells suppression in patients with Graves’ disease. Endocrine 2009, 36, 119–125. [Google Scholar] [CrossRef]
- Leskela, S.; Rodríguez-Muñoz, A.; De La Fuente, H.; Figueroa-Vega, N.; Bonay, P.; Martín, P.; Serrano, A.; Sánchez-Madrid, F.; González-Amaro, R.; Marazuela, M. Plasmacytoid dendritic cells in patients with autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 2013, 98, 2822–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weetman, A.P. Immunity, thyroid function and pregnancy: Molecular mechanisms. Nat. Rev. Endocrinol. 2010, 6, 311–318. [Google Scholar] [CrossRef]
- Coppola, A.; Tomasello, L.; Pitrone, M.; Cillino, S.; Richiusa, P.; Pizzolanti, G.; Giordano, C. Human limbal fibroblast-like stem cells induce immune-tolerance in autoreactive T lymphocytes from female patients with Hashimoto’s thyroiditis. Stem Cell Res. Ther. 2017, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braley-Mullen, H.; Sharp, G.C.; Medling, B.; Tang, H. Spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J. Autoimmun. 1999, 12, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, M.; Nagayama, Y.; Saitoh, O.; Sogawa, R.; Tone, S.; Abiru, N. Expression of immunoregulatory molecules by thyrocytes protects nonobese diabetic-H2h4 mice from developing autoimmune thyroiditis. Endocrinology 2009, 150, 1545–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, H.; Zhang, J.; Guo, Q.; Zhang, Y.; Zhong, X. Prunella vulgaris L. attenuates experimental autoimmune thyroiditis by inducing indoleamine 2,3-dioxygenase 1 expression and regulatory T cell expansion. Biomed. Pharmacother. 2020, 128, 110288. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Menicali, E.; Voce, P.; Morelli, S.; Cantarelli, S.; Sponziello, M.; Colella, R.; Fallarino, F.; Orabona, C.; Alunno, A.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) is up-regulated in thyroid carcinoma and drives the development of an immunosuppressant tumor microenvironment. J. Clin. Endocrinol. Metab. 2014, 99, E832–E840. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Neyaz, A.; Chougule, A.; Akita, M.; Zen, Y.; Forcione, D.; Castillo, C.F.; Ferrone, C.R.; Deshpande, V. Autoimmune Pancreatitis Type 2: Diagnostic Utility of PD-L1 Immunohistochemistry. Am. J. Surg. Pathol. 2019, 43, 898–906. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupa, A.; Kowalska, I. The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies. Int. J. Mol. Sci. 2021, 22, 9879. https://doi.org/10.3390/ijms22189879
Krupa A, Kowalska I. The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies. International Journal of Molecular Sciences. 2021; 22(18):9879. https://doi.org/10.3390/ijms22189879
Chicago/Turabian StyleKrupa, Anna, and Irina Kowalska. 2021. "The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies" International Journal of Molecular Sciences 22, no. 18: 9879. https://doi.org/10.3390/ijms22189879
APA StyleKrupa, A., & Kowalska, I. (2021). The Kynurenine Pathway—New Linkage between Innate and Adaptive Immunity in Autoimmune Endocrinopathies. International Journal of Molecular Sciences, 22(18), 9879. https://doi.org/10.3390/ijms22189879