N-Glycosylation as a Tool to Study Antithrombin Secretion, Conformation, and Function


 ,
,  , , and
, , and
Abstract
:1. Introduction
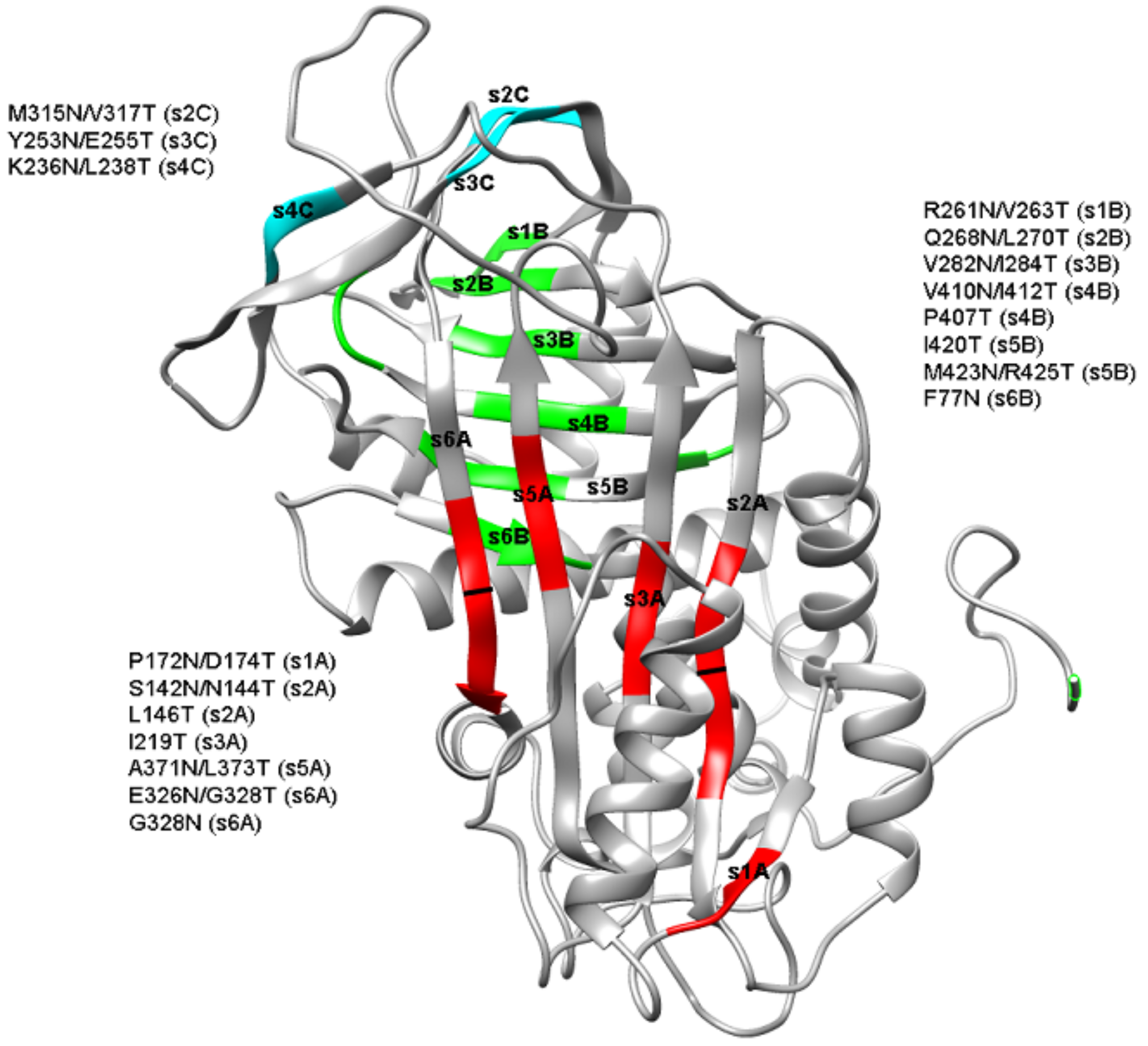
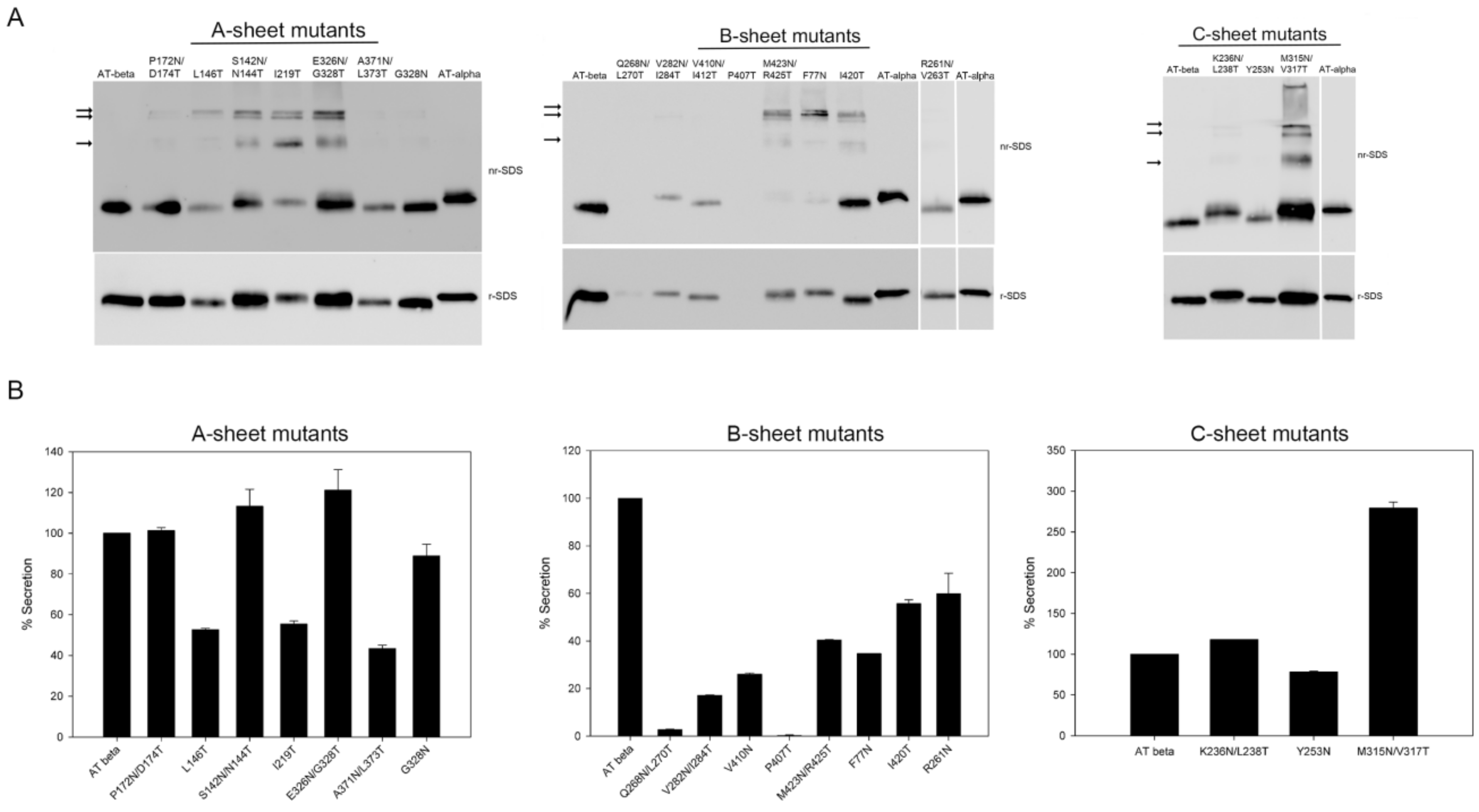
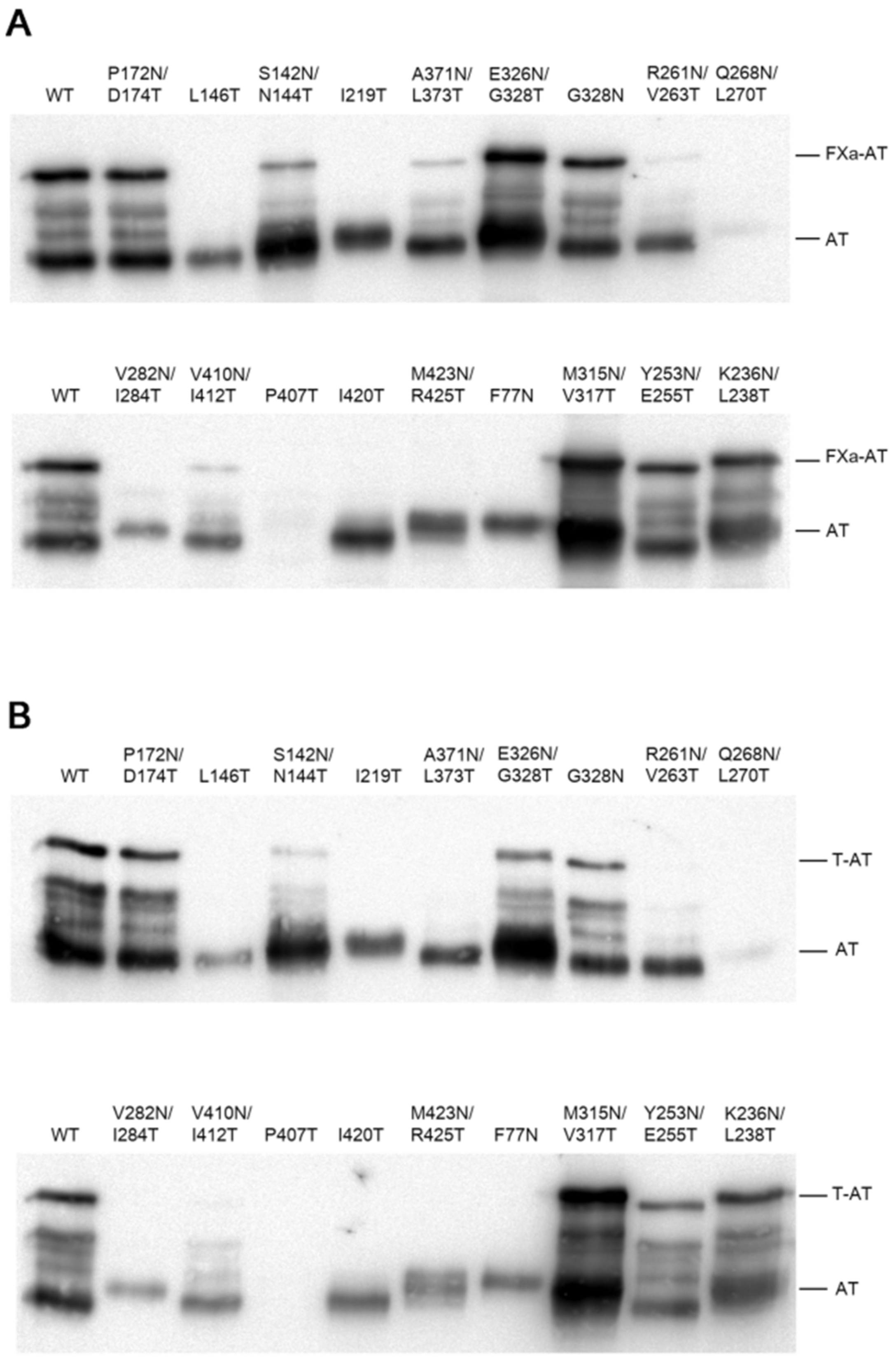
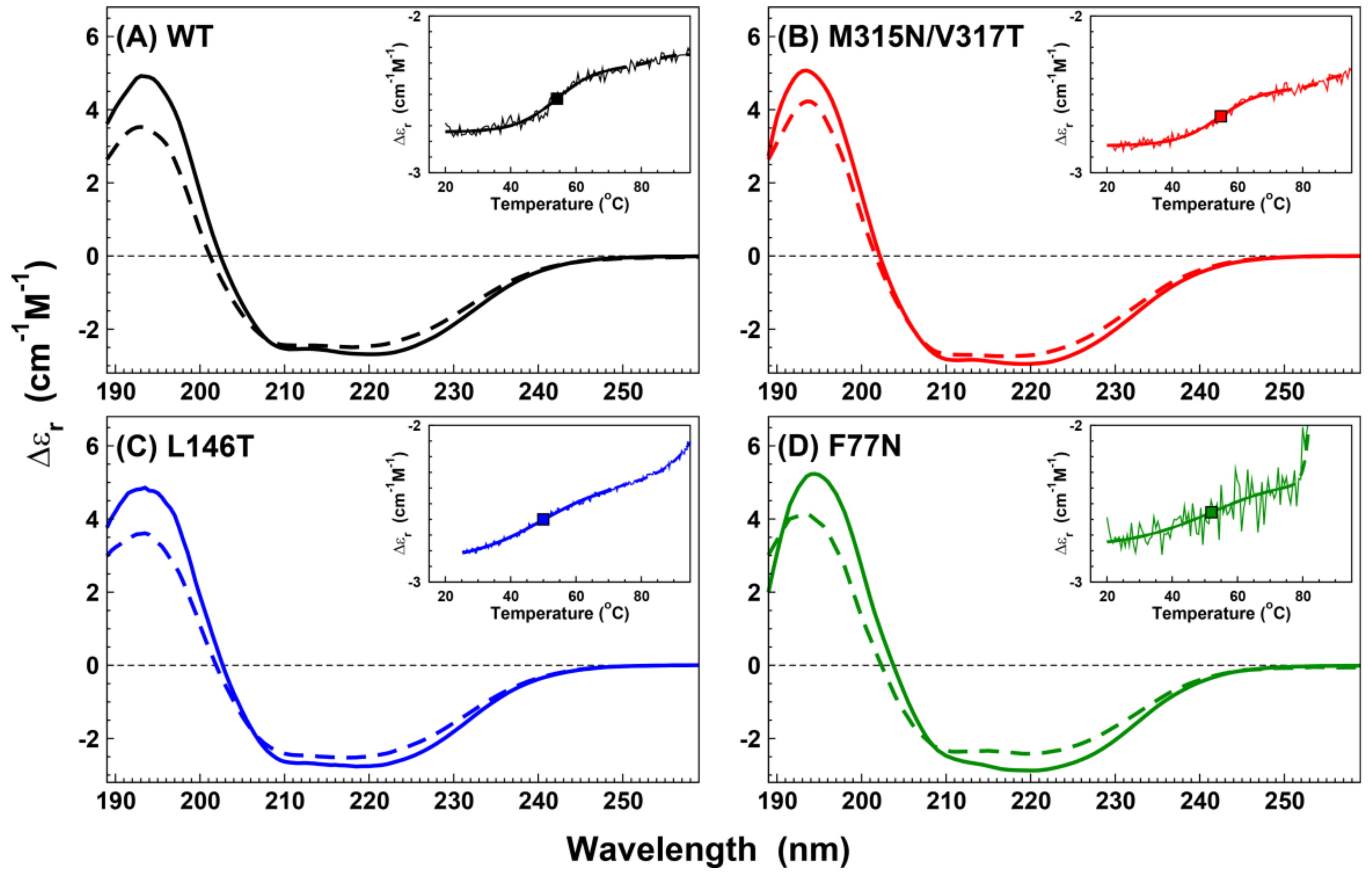
2. Results
2.1. Expression of Recombinant Mutants
2.2. Thrombin and FXa Complex Formation
2.3. Stability and Secondary Structure Determination
3. Discussion
4. Materials and Methods
4.1. Recombinant Expression of Wild Type and Mutant Antithrombins
4.2. Electrophoretic Analysis
4.3. Thrombin–Antithrombin and FXa-Antithrombin Complex Formation
4.4. Protein Purification
4.5. Circular Dichroism
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OST | Oligosaccharide transferase complex |
| CD | Circular dichroism |
| PDB | Protein Data Bank |
References
- Lijfering, W.M.; Brouwer, J.L.P.; Veeger, N.J.G.M.; Bank, I.; Coppens, M.; Middeldorp, S.; Hamulyák, K.; Prins, M.H.; Büller, H.R.; Van Der Meer, J. Selective testing for thrombophilia in patients with first venous thrombosis: Results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood 2009, 113, 5314–5322. [Google Scholar] [CrossRef] [Green Version]
- Wells, P.S.; Blajchman, M.A.; Henderson, P.; Wells, M.J.; Demers, C.; Bourque, R.; McAvoy, A. Prevalence of antithrombin deficiency in healthy blood donors: A cross-sectional study. Am. J. Hematol. 1994, 45, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Mulder, R.; Croles, F.N.; Mulder, A.B.; Huntington, J.A.; Meijer, K.; Lukens, M. V SERPINC1 gene mutations in antithrombin deficiency. Br. J. Haematol. 2017, 178, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Luxembourg, B.; Delev, D.; Geisen, C.; Spannagl, M.; Krause, M.; Miesbach, W.; Heller, C.; Bergmann, F.; Schmeink, U.; Grossmann, R.; et al. Molecular basis of antithrombin deficiency. Thromb. Haemost. 2011, 105, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushunje, A.; Evans, G.; Brennan, S.O.; Carrell, R.W.; Zhou, A. Latent antithrombin and its detection, formation and turnover in the circulation. J. Thromb. Haemost. 2004, 2, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Gettins, P.G.W.; Olson, S.T. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem. J. 2016, 473, 2273–2293. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.; Cosmi, F.; Ordoñez, A.; Berardelli, R.; Perez, J.; Guadagno, N.A.; Corda, L.; Marciniak, S.J.; Lomas, D.A.; Miranda, E. Polymers of Z α1-antitrypsin are secreted in cell models of disease. Eur. Respir. J. 2016, 47, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corral, J.; Huntington, J.A.; González-Conejero, R.; Mushunje, A.; Navarro, M.; Marco, P.; Vicente, V.; Carrell, R.W. Mutations in the shutter region of antithrombin result in formation of disulfide-linked dimers and severe venous thrombosis. J. Thromb. Haemost. 2004, 2, 931–939. [Google Scholar] [CrossRef]
- McCoy, A.J.; Pei, X.Y.; Skinner, R.; Abrahams, J.P.; Carrell, R.W. Structure of β-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J. Mol. Biol. 2003, 326, 823–833. [Google Scholar] [CrossRef]
- Bause, E. Structural requirements of N-glycosylation of proteins. Studies with proline peptides as conformational probes. Biochem. J. 1983, 209, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavel, Y.; von Heijne, G. Sequence differences between glycosylated and non-glycosylated asn-x-thr/ser acceptor sites: Implications for protein engineering. Protein Eng. Des. Sel. 1990, 3, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Shrimal, S.; Cherepanova, N.A.; Gilmore, R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 2015, 41, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pless, D.D.; Lennarz, W.J. Enzymatic conversion of proteins to glycoproteins. Proc. Natl. Acad. Sci. USA 1977, 74, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Águila, S.; Izaguirre, G.; Martínez-Martínez, I.; Vicente, V.; Olson, S.T.; Corral, J. Disease-causing mutations in the serpin antithrombin reveal a key domain critical for inhibiting protease activities. J. Biol. Chem. 2017, 292, 16513–16520. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, G.; Olson, S.T. Residues Tyr253 and Glu255 in strand 3 of beta-sheet C of antithrombin are key determinants of an exosite made accessible by heparin activation to promote rapid inhibition of factors Xa and IXa. J. Biol. Chem. 2006, 281, 13424–13432. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, G.; Aguila, S.; Qi, L.; Swanson, R.; Roth, R.; Rezaie, A.R.; Gettins, P.G.W.; Olson, S.T. Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J. Biol. Chem. 2014, 289, 34049–34064. [Google Scholar] [CrossRef] [Green Version]
- Noto, R.; Randazzo, L.; Raccosta, S.; Caccia, S.; Moriconi, C.; Miranda, E.; Martorana, V.; Manno, M. The stability and activity of human neuroserpin are modulated by a salt bridge that stabilises the reactive centre loop. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Noto, R.; Santangelo, M.G.; Ricagno, S.; Mangione, M.R.; Levantino, M.; Pezzullo, M.; Martorana, V.; Cupane, A.; Bolognesi, M.; Manno, M. The tempered polymerization of human neuroserpin. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Noto, R.; Santangelo, M.G.; Levantino, M.; Cupane, A.; Mangione, M.R.; Parisi, D.; Ricagno, S.; Bolognesi, M.; Manno, M.; Martorana, V. Functional and dysfunctional conformers of human neuroserpin characterized by optical spectroscopies and Molecular Dynamics. Biochim. Biophys. Acta - Proteins Proteomics 2015, 1854, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Knaupp, A.S.; Bottomley, S.P. Serpin polymerization and its role in disease - The molecular basis of α1-antitrypsin deficiency. IUBMB Life 2009, 61, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ekeowa, U.I.; Freeke, J.; Miranda, E.; Gooptu, B.; Bush, M.F.; Pérez, J.; Teckman, J.; Robinson, C.V.; Lomas, D.A. Defining the mechanism of polymerization in the serpinopathies. Proc. Natl. Acad. Sci. USA 2010, 107, 17146–17151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, M.; Sendall, T.J.; Harris, L.E.; Lewis, G.M.W.; Huntington, J.A. Loop-sheet mechanism of serpin polymerization tested by reactive center loop mutations. J. Biol. Chem. 2010, 285, 30752–30758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, M.; Li, W.; Johnson, D.J.D.; Huntington, J.A. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 2008, 455, 1255–1258. [Google Scholar] [CrossRef]
- Huntington, J.A.; Sendall, T.J.; Yamasaki, M. New insight into serpin polymerization and aggregation. Prion 2009, 3, 12–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordóñez, A.; Pérez, J.; Tan, L.; Dickens, J.A.; Motamedi-Shad, N.; Irving, J.A.; Haq, I.; Ekeowa, U.; Marciniak, S.J.; Miranda, E.; et al. A single-chain variable fragment intrabody prevents intracellular polymerization of Z α1-antitrypsin while allowing its antiproteinase activity. FASEB J. 2015, 29, 2667–2678. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, Y.; Dela Cruz, R.; Wintrode, P.L. Folding mechanism of the metastable serpin α 1-antitrypsin. Proc. Natl. Acad. Sci. USA 2012, 109, 4467–4472. [Google Scholar] [CrossRef] [Green Version]
- Lomas, D.A. Loop-sheet polymerization: The structural basis of Z α1-antitrypsin accumulation in the liver. Clin. Sci. 1994, 86, 489–495. [Google Scholar] [CrossRef]
- Lomas, D.A.; Mahadeva, R. α1-Antitrypsin polymerization and the serpinopathies: Pathobiology and prospects for therapy. J. Clin. Invest. 2002, 110, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.; Ferrarotti, I.; Berardelli, R.; Laffranchi, M.; Cerea, M.; Gangemi, F.; Haq, I.; Ottaviani, S.; Lomas, D.A.; Irving, J.A.; et al. The pathological Trento variant of alpha-1-antitrypsin (E75V) shows nonclassical behaviour during polymerization. FEBS J. 2017, 284, 2110–2126. [Google Scholar] [CrossRef] [PubMed]
- Kuwae, S.; Ohyama, M.; Ohya, T.; Ohi, H.; Kobayashi, K. Production of recombinant human antithrombin by Pichia pastoris. J. Biosci. Bioeng. 2005, 99, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Águila, S.; Martínez-Martínez, I.; Dichiara, G.; Gutiérrez-Gallego, R.; Navarro-Fernández, J.; Vicente, V.; Corral, J. Increased N-glycosylation efficiency by generation of an aromatic sequon on N135 of antithrombin. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Wang, F.; Orioli, S.; Ianeselli, A.; Spagnolli, G.; Beccara, S.A.; Gershenson, A.; Faccioli, P.; Wintrode, P.L. All-Atom Simulations Reveal How Single-Point Mutations Promote Serpin Misfolding. Biophys. J. 2018, 114, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Martínez, I.; Navarro-Fernández, J.; Aguila, S.; Miñano, A.; Bohdan, N.; De La Morena-Barrio, M.E.; Ordóñez, A.; Martínez, C.; Vicente, V.; Corral, J. The infective polymerization of conformationally unstable antithrombin mutants may play a role in the clinical severity of antithrombin deficiency. Mol. Med. 2012, 18, 762–770. [Google Scholar] [CrossRef]
- De la Morena-Barrio, M.; Sandoval, E.; Llamas, P.; Wypasek, E.; Toderici, M.; Navarro-Fernández, J.; Rodríguez-Alen, A.; Revilla, N.; López-Gálvez, R.; Miñano, A.; et al. High levels of latent antithrombin in plasma from patients with antithrombin deficiency. Thromb. Haemost. 2017, 117, 880–888. [Google Scholar] [CrossRef]
- Navarro-Fernández, J.; De La Morena-Barrio, M.E.; Padilla, J.; Miñano, A.; Bohdan, N.; Águila, S.; Martínez-Martínez, I.; Sevivas, T.S.; de Cos, C.; Fernández-Mosteirín, N.; et al. Antithrombin Dublin (p.Val30Glu): A relatively common variant with moderate thrombosis risk of causing transient antithrombin deficiency. Thromb. Haemost. 2016, 116, 146–154. [Google Scholar] [CrossRef]
- Mushunje, A.; Zhou, A.; Carrell, R.W.; Huntington, J.A. Heparin-induced substrate behavior of antithrombin Cambridge II. Blood 2003, 102, 4028–4034. [Google Scholar] [CrossRef]
- Irving, J.A.; Pike, R.N.; Lesk, A.M.; Whisstock, J.C. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 2000, 10, 1845–1864. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Martínez, I.; Johnson, D.J.D.; Yamasaki, M.; Navarro-Fernández, J.; Ordóñez, A.; Vicente, V.; Huntington, J.A.; Corral, J. Type II antithrombin deficiency caused by a large in-frame insertion: Structural, functional and pathological relevance. J. Thromb. Haemost. 2012, 10, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Martińez-Martínez, I.; Navarro-Fernández, J.; Østergaard, A.; Gutiérrez-Gallego, R.; Padilla, J.; Bohdan, N.; Miñano, A.; Pascual, C.; Martínez, C.; De La Morena-Barrio, M.E.; et al. Amelioration of the severity of heparin-binding antithrombin mutations by posttranslational mosaicism. Blood 2012, 120, 900–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norderman, B.; NYSTRöm, C.; BJörk, I. The Size and Shape of Human and Bovine Antithrombin III. Eur. J. Biochem. 1977, 78, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assigment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutants | Structure Location | Extra Glycan | Secretion Level | Anti FIIa Activity | Anti FXa Activity | Asn 3D Exposition |
|---|---|---|---|---|---|---|
| P172N/D174T | Strand 1A | NO | Normal | YES | YES | YES |
| L146T | Strand 2A | NO | Low | NO | NO | YES |
| S142N/N144T | Strand 2A | YES | Normal | YES | YES | NO |
| I219T | Strand 3A | YES | Low | NO | NO | NO |
| A371N/L373T | Strand 5A | NO | Low | NO | YES | NO |
| E326N/G328T | Strand 6A | YES | Normal | YES | YES | YES |
| G328N | Strand 6A | NO | Normal | YES | YES | YES |
| R261N/V263T | Strand 1B | NO | Low | NO | NO | YES |
| Q268N/L270T | Strand 2B | NO | Nil | NO | NO | YES |
| V282N/I284T | Strand 3B | YES | Very low | NO | NO | NO |
| V410N//I412T | Strand 4B | NO | Very low | NO | YES | NO |
| P407T | Beginning of strand 4B | NO | Nil | NO | NO | YES |
| I420T | Strand 5B | NO | Low | NO | NO | YES |
| M423N/R425T | Strand 5B | YES | Low | NO | NO | NO |
| F77N | Strand 6B | YES | Low | NO | NO | NO |
| M315N/V317T | Strand 2C | YES | Very high | YES | YES | YES |
| Y253N/E255T | Strand 3C | NO | Normal | YES | YES | YES |
| K236N/L238T | Strand 4C | YES | Normal | YES | YES | YES |
| Mutants | Alpha-Helix | Beta-Sheet | Turn | Coil |
|---|---|---|---|---|
| % | % | % | % | |
| WT | 27 ± 3 | 25 ± 3 | 17 ±4 | 31 ± 3 |
| M315N/V317T | 29 ± 2 | 22 ± 2 | 17 ± 4 | 32 ± 1 |
| L146T | 29 ± 2 | 23 ± 2 | 17 ± 4 | 31 ± 2 |
| F77N | 30 ± 2 | 23 ± 2 | 17 ± 4 | 30 ± 3 |
| pdb:1T1F | 28 | 27 | 25 | 20 |
| Mutation | Amino Acid Sequence | Presence and Position of Lysines | Extra N-Glycan |
|---|---|---|---|
| P172N/D174T | AKLQNLTFKEN | YES; K +4 | NO |
| L146T | LVSANRTFGDK | NO | NO |
| S142N/N144T | SKLVNATRLFG | YES; K−3 | YES |
| I219T | LVLVNTTYFHG | NO | YES |
| A371N/L373T | AFHKNFTEVNE | YES; K−1 | NO |
| E326N/G328T | RFRINDTFSLK | NO | YES |
| G328N | RIEDNFSLKEQ | YES; K + 4 | NO |
| R261N/V263T | KFRYNRTAEGT | YES; K − 4 | NO |
| Q268N/L270T | AEGTNVTELPF | NO | YES |
| V282N/I284T | DITMNLTLPKP | NO | YES |
| V410N/I412T | RPFLNFTREVP | NO | NO |
| P407T | TFKANRTFLVF | YES; K − 2 | NO |
| I420T | EVPLNTTIFMG | NO | NO |
| M423N/R425T | TIIFNGTVANP | NO | YES |
| F77N | NDNINLSPLSI | NO | YES |
| M315N/V317T | LEEMNLTVHMP | NO | YES |
| Y253N/E255T | ASMMNQTGKFR | YES; K + 4 | NO |
| K236N/L238T | ENTRNETFYKA | NO | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Águila, S.; Noto, R.; Luengo-Gil, G.; Espín, S.; Bohdan, N.; de la Morena-Barrio, M.E.; Peñas, J.; Rodenas, M.C.; Vicente, V.; Corral, J.; et al. N-Glycosylation as a Tool to Study Antithrombin Secretion, Conformation, and Function. Int. J. Mol. Sci. 2021, 22, 516. https://doi.org/10.3390/ijms22020516
Águila S, Noto R, Luengo-Gil G, Espín S, Bohdan N, de la Morena-Barrio ME, Peñas J, Rodenas MC, Vicente V, Corral J, et al. N-Glycosylation as a Tool to Study Antithrombin Secretion, Conformation, and Function. International Journal of Molecular Sciences. 2021; 22(2):516. https://doi.org/10.3390/ijms22020516
Chicago/Turabian StyleÁguila, Sonia, Rosina Noto, Ginés Luengo-Gil, Salvador Espín, Nataliya Bohdan, María Eugenia de la Morena-Barrio, Julia Peñas, Maria Carmen Rodenas, Vicente Vicente, Javier Corral, and et al. 2021. "N-Glycosylation as a Tool to Study Antithrombin Secretion, Conformation, and Function" International Journal of Molecular Sciences 22, no. 2: 516. https://doi.org/10.3390/ijms22020516
APA StyleÁguila, S., Noto, R., Luengo-Gil, G., Espín, S., Bohdan, N., de la Morena-Barrio, M. E., Peñas, J., Rodenas, M. C., Vicente, V., Corral, J., Manno, M., & Martínez-Martínez, I. (2021). N-Glycosylation as a Tool to Study Antithrombin Secretion, Conformation, and Function. International Journal of Molecular Sciences, 22(2), 516. https://doi.org/10.3390/ijms22020516