Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction

 ,
,  ,
,  ,
,  and
and
Abstract
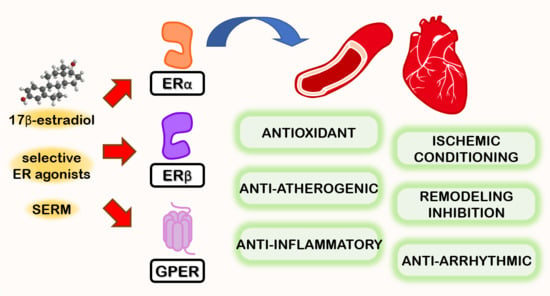
:
1. Introduction
2. Estrogen Receptors and Distribution in the Cardiovascular System
3. General Mechanism of Action of ER
4. ER Modulation and MI: Preclinical Studies
5. ER Modulation and MI: Clinical Studies
6. Genetic Factors Related to Estrogen Receptors and MI
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2016 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1260–1344. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, W.S.; Daniels, S.R.; Burke, L.E.; Franklin, B.A.; Goff, D.C.; Hayman, L.L.; Lloyd-Jones, D.; Pandey, D.K.; Sanchez, E.J.; Schram, A.P.; et al. Value of Primordial and Primary Prevention for Cardiovascular Disease. Circulation 2011, 124, 967–990. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart J. 2014, 35, 2929–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American college of cardiology/American heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2019, 74, e177–e232. [Google Scholar] [CrossRef]
- Ibáñez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur. Heart J. 2017, 39, 119–177. [Google Scholar] [CrossRef] [Green Version]
- Aimo, A.; Vergaro, G.; Barison, A.; Maffei, S.; Borrelli, C.; Morrone, D.; Cameli, M.; Palazzuoli, A.; Ambrosio, G.; Coiro, S.; et al. Sex-related differences in chronic heart failure. Int. J. Cardiol. 2018, 255, 145–151. [Google Scholar] [CrossRef]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2020. Available online: https://onlinelibrary.wiley.com/doi/10.1002/ehf2.13144 (accessed on 21 December 2020).
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Barton, M. Estrogens and Coronary Artery Disease: New Clinical Perspectives, 1st ed.; Elsevier Inc.: Cambridge, MA, USA, 2016; Volume 77, ISBN 9780128043967. [Google Scholar]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Prabhushankar, R.; Krueger, C.; Manrique-Acevedo, C. Membrane Estrogen Receptors: Their Role in Blood Pressure Regulation and Cardiovascular Disease. Curr. Hypertens. Rep. 2014, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzaneh, S.; Zarghi, A. Estrogen Receptor Ligands: A Review (2013–2015). Sci. Pharm. 2016, 84, 409. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodzadeh, S.; Dworatzek, E. The Role of 17β-Estradiol and Estrogen Receptors in Regulation of Ca2+ Channels and Mitochondrial Function in Cardiomyocytes. Front. Endocrinol. 2019, 10, 310. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Piquereau, J.; Veksler, V.; Garnier, A. Estrogens, Estrogen Receptors Effects on Cardiac and Skeletal Muscle Mitochondria. Front. Endocrinol. 2019, 10, 557. [Google Scholar] [CrossRef] [Green Version]
- Otto, C.; Fuchs, I.; Kauselmann, G.; Kern, H.; Zevnik, B.; Andreasen, P.; Schwarz, G.; Altmann, H.; Klewer, M.; Schoor, M.; et al. GPR30 Does Not Mediate Estrogenic Responses in Reproductive Organs in Mice. Biol. Reprod. 2009, 80, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Isensee, J.; Meoli, L.; Zazzu, V.; Nabzdyk, C.; Witt, H.; Soewarto, D.; Effertz, K.; Fuchs, H.; Gailus-Durner, V.; Busch, D.; et al. Expression Pattern of G Protein-Coupled Receptor 30 in LacZ Reporter Mice. Endocrinology 2008, 150, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Randeva, H.S.; Patel, V.H.; Chen, J.; Ramanjaneya, M.; Karteris, E.; Zachariades, E.; Thomas, P.; Been, M. G-protein coupled estrogen receptor 1 expression in rat and human heart: Protective role during ischaemic stress. Int. J. Mol. Med. 2010, 26, 193–199. [Google Scholar] [CrossRef]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.F.; Bopassa, J.C. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3β Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef] [Green Version]
- Bopassa, J.C.; Eghbali, M.; Toro, L.; Stefani, E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H16–H23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G Protein-coupled Estrogen Receptor Protects from Atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Madungwe, N.B.; da Cruz JunHo, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Korach, K.S. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Pedram, A.; Razandi, M.; O’Mahony, F.; Lubahn, D.; Levin, E.R. Estrogen Receptor-β Prevents Cardiac Fibrosis. Mol. Endocrinol. 2010, 24, 2152–2165. [Google Scholar] [CrossRef] [Green Version]
- Levin, E.R. Membrane estrogen receptors signal to determine transcription factor function. Steroids 2018, 132, 1–4. [Google Scholar] [CrossRef]
- Menazza, S.; Murphy, E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef]
- Patten, R.D.; Karas, R.H. Estrogen Replacement and Cardiomyocyte Protection. Trends Cardiovasc. Med. 2006, 16, 69–75. [Google Scholar] [CrossRef]
- Meyer, M.R.; Baretella, O.; Prossnitz, E.R.; Barton, M. Dilation of Epicardial Coronary Arteries by the G Protein-Coupled Estrogen Receptor Agonists G-1 and ICI 182,780. Pharmacology 2010, 86, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, S.H.; Cohen, J.A.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Chronic Treatment with the G Protein-Coupled Receptor 30 Agonist G-1 Decreases Blood Pressure in Ovariectomized mRen2.Lewis Rats. Endocrinology 2009, 150, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Amann, K.; Field, A.S.; Hu, C.; Hathaway, H.J.; Kanagy, N.L.; Walker, M.K.; Barton, M.; Prossnitz, E.R. Deletion of G Protein–Coupled Estrogen Receptor Increases Endothelial Vasoconstriction. Hypertension 2012, 59, 507–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, E.; Bhattacharya, I.; Brailoiu, E.; Damjanović, M.; Brailoiu, G.C.; Gao, X.; Mueller-Guerre, L.; Marjon, N.A.; Gut, A.; Minotti, R.; et al. Regulatory Role of G Protein–Coupled Estrogen Receptor for Vascular Function and Obesity. Circ. Res. 2009, 104, 288–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesson, C.S.; Sahlin, L. Expression pattern and signalling pathways in neutrophil like HL-60 cells after treatment with estrogen receptor selective ligands. Mol. Cell. Endocrinol. 2012, 361, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Z.; Lin, M.; Groban, L. Activation of GPR30 inhibits cardiac fibroblast proliferation. Mol. Cell. Biochem. 2015, 405, 135–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Suzuki, T.; Mizukami, Y.; Ikeda, T. The membrane-type estrogen receptor G-protein-coupled estrogen receptor suppresses lipopolysaccharide-induced interleukin 6 via inhibition of nuclear factor-kappa B pathway in murine macrophage cells. Anim. Sci. J. 2017, 88, 1870–1879. [Google Scholar] [CrossRef]
- Sack, M.; Rader, D.J.; Cannon, R. Oestrogen and inhibition of oxidation of low-density lipoproteins in postmenopausal women. Lancet 1994, 343, 269–270. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Yang, X.-P.; Liu, Y.-H.; Mehta, D.; Karumanchi, R.; Bulagannawar, M.; Carretero, O.A. Effects of ACE Inhibitor, AT1 Antagonist, and Combined Treatment in Mice with Heart Failure. J. Cardiovasc. Pharmacol. 2000, 36, 472–480. [Google Scholar] [CrossRef]
- Van Eickels, M.; Patten, R.D.; Aronovitz, M.J.; Alsheikh-Ali, A.; Gostyla, K.; Celestin, F.; Grohe, C.; Mendelsohn, M.E.; Karas, R.H. 17-Beta-Estradiol increases cardiac remodeling and mortality in mice with myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 2084–2092. [Google Scholar] [CrossRef]
- Smith, P.J.W.; Ornatsky, O.; Stewart, D.J.; Picard, P.; Dawood, F.; Wen, W.-H.; Liu, P.P.; Webb, D.J.; Monge, J.C. Effects of estrogen replacement on infarct size, cardiac remodeling, and the endothelin system after myocardial infarction in ovariectomized rats. Circulation 2000, 102, 2983–2989. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-H.; Su, S.-F.; Chou, T.-F.; Lee, T. Differential Effects of Sarcolemmal and Mitochondrial KATP Channels Activated by 17β-Estradiol on Reperfusion Arrhythmias and Infarct Sizes in Canine Hearts. J. Pharmacol. Exp. Ther. 2002, 301, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Patten, R.D.; Pourati, I.; Aronovitz, M.J.; Baur, J.; Celestin, F.; Chen, X.; Michael, A.; Haq, S.; Nuedling, S.; Grohe, C.; et al. 17β-Estradiol Reduces Cardiomyocyte Apoptosis In Vivo and In Vitro via Activation of Phospho-Inositide-3 Kinase/Akt Signaling. Circ. Res. 2004, 95, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, T.; Neumann, M.; De Jager, T.; Jazbutytea, V.; Neysesa, L. Estrogen Effects in the Myocardium: Inhibition of NF-κB DNA Binding by Estrogen Receptor-α and -β. Biochem. Biophys. Res. Commun. 2001, 286, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, T.; Schumann, M.; Neumann, M.; Dejager, T.; Stimpel, M.; Serfling, E.; Neyses, L. 17β-Estradiol Prevents Programmed Cell Death in Cardiac Myocytes. Biochem. Biophys. Res. Commun. 2000, 268, 192–200. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Levin, E.R. Estrogen Inhibits Cardiomyocyte Hypertrophyin Vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP. J. Biol. Chem. 2005, 280, 26339–26348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Gou, Y.; Zhang, H.; Zuo, H.; Zhang, H.; Liu, Z.-X.; Yao, D. Estradiol improves cardiovascular function through up-regulation of SOD2 on vascular wall. Redox. Biol. 2014, 3, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, H.; Kim, M.K.; Iwakura, A.; Ii, M.; Thorne, T.; Qin, G.; Asai, J.; Tsutsumi, Y.; Sekiguchi, H.; Silver, M.; et al. Estrogen Receptors α and β Mediate Contribution of Bone Marrow–Derived Endothelial Progenitor Cells to Functional Recovery After Myocardial Infarction. Circulation 2006, 114, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Lin, C.-C.; Lee, T. 17β-estradiol decreases vulnerability to ventricular arrhythmias by preserving Connexin43 protein in infarcted rats. Eur. J. Pharmacol. 2010, 629, 73–81. [Google Scholar] [CrossRef]
- Yuan, Z.; Kang, L.; Wang, Z.; Chen, A.; Zhao, Q.; Li, H. 17β-estradiol promotes recovery after myocardial infarction by enhancing homing and angiogenic capacity of bone marrow-derived endothelial progenitor cells through ERα-SDF-1/CXCR4 crosstalking. Acta Biochim. Biophys. Sin. 2018, 50, 1247–1256. [Google Scholar] [CrossRef]
- Lee, T.; Lin, S.-Z.; Chang, N.-C. Membrane ERα attenuates myocardial fibrosis via RhoA/ROCK-mediated actin remodeling in ovariectomized female infarcted rats. J. Mol. Med. 2013, 92, 43–51. [Google Scholar] [CrossRef]
- Nikolic, I.; Liu, D.; Bell, J.A.; Collins, J.; Steenbergen, C.; Murphy, E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J. Mol. Cell. Cardiol. 2007, 42, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Steenbergen, C.; Murphy, E.; Sun, J. Estrogen Receptor-β Activation Results in S-Nitrosylation of Proteins Involved in Cardioprotection. Circulation 2009, 120, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedram, A.; Razandi, M.; Narayanan, R.; Levin, E.R. Estrogen receptor beta signals to inhibition of cardiac fibrosis. Mol. Cell. Endocrinol. 2016, 434, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Booth, E.A.; Marchesi, M.; Knittel, A.K.; Kilbourne, E.J.; Lucchesi, B.R. The Pathway-Selective Estrogen Receptor Ligand WAY-169916 Reduces Infarct Size after Myocardial Ischemia and Reperfusion by an Estrogen Receptor Dependent Mechanism. J. Cardiovasc. Pharmacol. 2007, 49, 401–407. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Ning, H.; Na, L.; Niu, Y.; Wang, M.; Feng, R.; Liu, L.; Guo, F.; Hou, S.; et al. Calcium supplementation increases circulating cholesterol by reducing its catabolism via GPER and TRPC1-dependent pathway in estrogen deficient women. Int. J. Cardiol. 2013, 168, 2548–2560. [Google Scholar] [CrossRef]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.M.; Lin, S.Z.; Chang, N.C. Both GPER and membrane oestrogen receptor-α activation protect ventricular remodelling in 17β oestradiol-treated ovariectomized infarcted rats. J. Cell. Mol. Med. 2014, 18, 2454–2465. [Google Scholar] [CrossRef]
- Wang, X.; Lu, L.; Tan, Y.; Jiang, L.; Zhao, M.; Gao, E.; Yu, S.; Liu, J. GPR 30 reduces myocardial infarct area and fibrosis in female ovariectomized mice by activating the PI3K/AKT pathway. Life Sci. 2019, 226, 22–32. [Google Scholar] [CrossRef]
- Zhan, Y.; Liu, Z.; Li, M.; Ding, T.; Zhang, L.; Lu, Q.; Liu, X.; Zhang, Z.; Vlessidis, A.; Aw, T.Y.; et al. ERβ expression in the endothelium ameliorates ischemia/reperfusion-mediated oxidative burst and vascular injury. Free. Radic. Biol. Med. 2016, 96, 223–233. [Google Scholar] [CrossRef]
- Zhang, J.-B.; Guo, C.-L. Protective effect and mechanism of estrogen receptor β on myocardial infarction in mice. Exp. Ther. Med. 2017, 14, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Gabel, S.A.; Walker, V.R.; London, R.E.; Steenbergen, C.; Korach, K.S.; Murphy, E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Lubahn, D.B.; Liu, J.; Vannan, M.; Levin, E.R. Estrogen Inhibits Cardiac Hypertrophy: Role of Estrogen Receptor-β to Inhibit Calcineurin. Endocrinology 2008, 149, 3361–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wang, Y.; Weil, B.; Abarbanell, A.; Herrmann, J.L.; Tan, J.; Kelly, M.; Meldrum, D.R. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R972–R978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, T.; Fuchs, M.; Arkudas, A.; Geertz, S.; Meyer, R.; Gardiwal, A.; Klein, G.; Niehaus, M.; Krust, A.; Chambon, P.; et al. Female Mice Lacking Estrogen Receptor β Display Prolonged Ventricular Repolarization and Reduced Ventricular Automaticity after Myocardial Infarction. Circulation 2005, 111, 2282–2290. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Fredette, N.C.; Barton, M.; Prossnitz, E.R. G protein-coupled estrogen receptor inhibits vascular prostanoid production and activity. J. Endocrinol. 2015, 227, 61–69. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, S.A.; Claudio, E.R.G.; Mengal, V.; Brasil, G.A.; Merlo, E.; Podratz, P.L.; Graceli, J.B.; Gouvea, S.A.; De Abreu, G.R. Estrogen Therapy Worsens Cardiac Function and Remodeling and Reverses the Effects of Exercise Training After Myocardial Infarction in Ovariectomized Female Rats. Front. Physiol. 2018, 9, 1242. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Xiao, D.; Zhang, L. Estrogen Regulates Angiotensin II Receptor Expression Patterns and Protects the Heart from Ischemic Injury in Female Rats1. Biol. Reprod. 2015, 93, 6. [Google Scholar] [CrossRef] [Green Version]
- Babiker, F.A.; Lips, D.J.; Delvaux, E.; Zandberg, P.; Janssen, B.J.A.; Prinzen, F.; Van Eys, G.; Grohé, C.; Doevendans, P.A. Oestrogen modulates cardiac ischaemic remodelling through oestrogen receptor-specific mechanisms. Acta Physiol. 2007, 189, 23–31. [Google Scholar] [CrossRef]
- Veeneman, G.H. Subtype-Selective Estrogens. In Nuclear Receptors as Drug Targets; Ottow, E., Weinmann, H., Eds.; Wiley-VCH Verlag: Verlag/Berlin, Germany, 2008; Volume 39, pp. 65–126. ISBN 9783527623297. [Google Scholar]
- Gaudet, H.; Cheng, S.; Christensen, E.; Filardo, E.J. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol. Cell. Endocrinol. 2015, 418, 207–219. [Google Scholar] [CrossRef]
- Vehkavaara, S.; Hakala-Ala-Pietilä, T.; Virkamäki, A.; Bergholm, R.; Ehnholm, C.; Hovatta, O.; Taskinen, M.-R.; Yki-Järvinen, H. Differential Effects of Oral and Transdermal Estrogen Replacement Therapy on Endothelial Function in Postmenopausal Women. Circulation 2000, 102, 2687–2693. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Jin, X.; Zeng, Z.; Liu, W.; Wang, B.; Wang, H. Estrogen-replacement therapy promotes angiogenesis after acute myocardial infarction by enhancing SDF-1 and estrogen receptor expression. Microvasc. Res. 2009, 77, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Broberg, A.M.; Siddiqui, A.J.; Fischer, H.; Grinnemo, K.-H.; Wärdell, E.; Andersson, A.B.; Inzunza, J.; Sylvén, C.; Gustafsson, J.Å. Estrogen receptors do not influence angiogenesis after myocardial infarction. Scand. Cardiovasc. J. 2011, 45, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Nanao-Hamai, M.; Son, B.-K.; Hashizume, T.; Ogawa, S.; Akishita, M. Protective effects of estrogen against vascular calcification via estrogen receptor α-dependent growth arrest-specific gene 6 transactivation. Biochem. Biophys. Res. Commun. 2016, 480, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Zhang, W.; Liu, J. Membrane estrogen receptor alpha is an important modulator of bone marrow C-Kit+ cells mediated cardiac repair after myocardial infarction. Int. J. Clin. Exp. Pathol. 2015, 8, 4284–4295. [Google Scholar]
- Brinckmann, M.; Kaschina, E.; Altarche-Xifró, W.; Curato, C.; Timm, M.; Grzesiak, A.; Dong, J.; Kappert, K.; Kintscher, U.; Unger, T.; et al. Estrogen receptor α supports cardiomyocytes indirectly through post-infarct cardiac c-kit+ cells. J. Mol. Cell. Cardiol. 2009, 47, 66–75. [Google Scholar] [CrossRef]
- Darwesh, A.M.; El-Azab, M.F.; Abo-Gresha, N.M.; El-Sayed, N.M.; Moustafa, Y.M. Cardioprotective Mechanisms of Exenatide in Isoprenaline-induced Myocardial Infarction. J. Cardiovasc. Pharmacol. 2018, 71, 160–173. [Google Scholar] [CrossRef]
- Duft, K.; Schanz, M.; Pham, H.; Abdelwahab, A.; Schriever, C.; Kararigas, G.; Dworatzek, E.; Davidson, M.M.; Regitz-Zagrosek, V.; Morano, I.; et al. 17β-Estradiol-induced interaction of estrogen receptor α and human atrial essential myosin light chain modulates cardiac contractile function. Basic Res. Cardiol. 2016, 112, 1. [Google Scholar] [CrossRef]
- Huang, P.-C.; Kuo, W.; Shen, C.-Y.; Chen, Y.-F.; Lin, Y.-M.; Ho, T.-J.; Padma, V.V.; Lo, J.-F.; Huang, C. Anthocyanin Attenuates Doxorubicin-Induced Cardiomyotoxicity via Estrogen Receptor-α/β and Stabilizes HSF1 to Inhibit the IGF-IIR Apoptotic Pathway. Int. J. Mol. Sci. 2016, 17, 1588. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Shan, J.; Feng, A.; Schmull, S.; Gu, J.; Xue, S. Oestrogen Receptor β Activation Protects Against Myocardial Infarction via Notch1 Signalling. Cardiovasc. Drugs Ther. 2020, 34, 165–178. [Google Scholar] [CrossRef]
- Kusama, M.; Miyauchi, K.; Aoyama, H.; Sano, M.; Kimura, M.; Mitsuyama, S.; Komaki, K.; Doihara, H. Effects of toremifene (TOR) and tamoxifen (TAM) on serum lipids in postmenopausal patients with breast cancer. Breast Cancer Res. Treat. 2004, 88, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.; Gou, Y.; Yu, H.; Siminelakis, S.; Wang, S.; Kong, D.; Zhou, Y.; Liu, Z.-X.; Ding, Y.; et al. Estradiol mediates vasculoprotection via ERRα-dependent regulation of lipid and ROS metabolism in the endothelium. J. Mol. Cell. Cardiol. 2015, 87, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Morgan, M.; Shen, R.-F.; Steenbergen, C.; Murphy, E. Preconditioning Results in S-Nitrosylation of Proteins Involved in Regulation of Mitochondrial Energetics and Calcium Transport. Circ. Res. 2007, 101, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Medzikovic, L.; Aryan, L.; Eghbali, M. Connecting sex differences, estrogen signaling, and microRNAs in cardiac fibrosis. J. Mol. Med. 2019, 97, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Hass, C.; Panda, B.P.; Khanam, R.; Najmi, A.K.; Akhtar, M. Histamine H3 Receptor Agonist Imetit Attenuated Isoproterenol Induced Renin Angiotensin System and Sympathetic Nervous System Overactivity in Myocardial Infarction of Rats. Drug Res. 2016, 66, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Okuda, T.; Shirai, K.; Urata, H. Increase of chymase-dependent angiotensin II-forming activity in circulating mononuclear leukocytes after acute myocardial infarction chymase activity after acute myocardial infarction. Heart Vessel. 2019, 34, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.S.; Gabriel-Costa, D.; Wang, H.; Ahmad, S.; Sun, X.; Varagic, J.; Sudo, R.T.; Ferrario, C.M.; Dell Italia, L.J.D.; Zapata-Sudo, G.; et al. Blunting of cardioprotective actions of estrogen in female rodent heart linked to altered expression of cardiac tissue chymase and ACE2. J. Renin-Angiotensin-Aldosterone Syst. 2017, 18, 147032031772227. [Google Scholar] [CrossRef] [Green Version]
- Tomicek, N.J.; Miller-Lee, J.L.; Hunter, J.C.; Korzick, N.H. Estrogen Receptor Beta Does Not Influence Ischemic Tolerance in the Aged Female Rat Heart. Cardiovasc. Ther. 2011, 31, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Schuster, I.; Mahmoodzadeh, S.; Dworatzek, E.; Jaisser, F.; Messaoudi, S.; Morano, I.; Regitz-Zagrosek, V. Cardiomyocyte-specific overexpression of oestrogen receptor β improves survival and cardiac function after myocardial infarction in female and male mice. Clin. Sci. 2016, 130, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-Y.; Chen, J.-S.; Wu, X.-B.; Shyu, W.-C.; Chaunchaiyakul, R.; Zhao, X.-L.; Kuo, C.-H.; Cheng, Y.-J.; Yang, A.-L.; Lee, S.-D. Combined effects of 17β-estradiol and exercise training on cardiac apoptosis in ovariectomized rats. PLoS ONE 2018, 13, e0208633. [Google Scholar] [CrossRef]
- Bulut, E.C.; Abueid, L.; Ercan, F.; Süleymanoğlu, S.; Ağırbaşlı, M.; Yeğen, B.Ç. Treatment with oestrogen-receptor agonists or oxytocin in conjunction with exercise protects against myocardial infarction in ovariectomized rats. Exp. Physiol. 2016, 101, 612–627. [Google Scholar] [CrossRef]
- Hussain, Y.; Ding, Q.; Connelly, P.W.; Brunt, J.H.; Ban, M.R.; McIntyre, A.D.; Huff, M.W.; Gros, R.; Hegele, R.A.; Feldman, R.D. G-Protein Estrogen Receptor as a Regulator of Low-Density Lipoprotein Cholesterol Metabolism. Arter. Thromb. Vasc. Biol. 2015, 35, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvin, A.M.; Jackson, M.A.; Korzick, D.H. Inhibition of programmed necrosis limits infarct size through altered mitochondrial and immune responses in the aged female rat heart. Am. J. Physiol. Circ. Physiol. 2018, 315, H1434–H1442. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, V.; Wauson, E.; Christian, D.; Clayton, S.; Giles, J.; Tran, Q.-K. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem. Pharmacol. 2020, 171, 113727. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, S.J.; Chou, T.M.; Chatterjee, K.; Sudhir, K. Tamoxifen Is an Acute, Estrogen-like, Coronary Vasodilator of Porcine Coronary Arteries In Vitro. J. Cardiovasc. Pharmacol. 2001, 38, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Tao, Y.; Chen, M.; Yu, J.; Li, W.-J.; Tao, L.; Li, Y.; Li, F. 17β-Estradiol Promotes Angiogenesis of Rat Cardiac Microvascular Endothelial Cells In Vitro. Med Sci. Monit. 2018, 24, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Rayabarapu, N.; Patel, B.M. Beneficial role of tamoxifen in isoproterenol-induced myocardial infarction. Can. J. Physiol. Pharmacol. 2014, 92, 849–857. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Protein–Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.-T.; Cheng, P.-Y.; Lam, K.-K.; Chen, S.-Y.; Ting, Y.-F.; Yen, M.-H.; Lee, Y.-M. Cardioprotective effects of long-term treatment with raloxifene, a selective estrogen receptor modulator, on myocardial ischemia/reperfusion injury in ovariectomized rats. Menopause 2010, 17, 127–134. [Google Scholar] [CrossRef]
- Chan, Y.-C.; Leung, F.P.; Tian, X.Y.; Yung, L.M.; Lau, C.W.; Chen, Z.-Y.; Yao, X.; Laher, I.; Huang, Y. Raloxifene improves vascular reactivity in pressurized septal coronary arteries of ovariectomized hamsters fed cholesterol diet. Pharmacol. Res. 2012, 65, 182–188. [Google Scholar] [CrossRef]
- Pósa, A.; Szabó, R.; Kupai, K.; Berkó, A.M.; Veszelka, M.; Szűcs, G.; Börzsei, D.; Gyöngyösi, M.; Pávó, I.; Deim, Z.; et al. Cardioprotective Effect of Selective Estrogen Receptor Modulator Raloxifene Are Mediated by Heme Oxygenase in Estrogen-Deficient Rat. Oxidative Med. Cell. Longev. 2017, 2017, 2176749. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F.; Merz, N.B.; Barnes, P.J.; Brinton, R.D.; Carrero, J.-J.; DeMeo, D.L.; De Vries, G.J.; Epperson, C.N.; Govindan, R.; Klein, S.L.; et al. Sex and gender: Modifiers of health, disease, and medicine. Lancet 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Mehta, L.S.; Beckie, T.M.; Devon, H.A.; Grines, C.L.; Krumholz, H.M.; Johnson, M.N.; Lindley, K.J.; Vaccarino, V.; Wang, T.Y.; Watson, K.E.; et al. Acute Myocardial Infarction in Women. Circulation 2016, 133, 916–947. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, M.C.; Zekavat, S.M.; Aragam, K.; Finneran, P.; Klarin, D.; Bhatt, D.L.; Januzzi, J.L.; Scott, N.S.; Natarajan, P. Association of Premature Natural and Surgical Menopause with Incident Cardiovascular Disease. JAMA 2019, 322, 2411. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dustin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Marko, K.I.; Simon, J.A. Clinical trials in menopause. Menopause 2018, 25, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Appiah, D.; Schreiner, P.J.; Demerath, E.W.; Loehr, L.R.; Chang, P.P.; Folsom, A.R. Association of Age at Menopause with Incident Heart Failure: A Prospective Cohort Study and Meta-Analysis. J. Am. Heart Assoc. 2016, 5, e003769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, B.K.; Knutsen, S.F.; Fraser, G.E. Age at Natural Menopause and Total Mortality and Mortality from Ischemic Heart Disease. J. Clin. Epidemiol. 1999, 52, 303–307. [Google Scholar] [CrossRef]
- Paciuc, J. Hormone Therapy in Menopause. In Hormonal Pathology of the Uterus. Advances in Experimental Medicine and Biology; Deligdisch-Schor, L., Miceli, A.M., Eds.; Springer: Cham, Switzerland, 2020; Volume 1242, pp. 89–120. [Google Scholar]
- Okoth, K.; Chandan, J.S.; Marshall, T.; Thangaratinam, S.; Thomas, G.N.; Nirantharakumar, K.; Adderley, N.J. Association between the reproductive health of young women and cardiovascular disease in later life: Umbrella review. BMJ 2020, m3502. [Google Scholar] [CrossRef]
- Canpolat, U.; Tokgozoglu, L.; Yorgun, H.; Kaya, E.B.; Gürses, K.M.; Sahiner, L.; Bozdağ, G.; Kabakçi, G.; Oto, A.; Aytemir, K. The association of premature ovarian failure with ventricular repolarization dynamics evaluated by QT dynamicity. Europace 2013, 15, 1657–1663. [Google Scholar] [CrossRef]
- Aksoy, M.N.M.; Akdemir, N.; Kilic, H.; Yilmaz, S.; Akdemir, N.; Gunduz, H. Pulse wave velocity and myocardial performance index in premature ovarian insufficiency. Scand. Cardiovasc. J. 2017, 51, 95–98. [Google Scholar] [CrossRef]
- Podfigurna-Stopa, A.; Czyzyk, A.; Grymowicz, M.; Smolarczyk, R.; Katulski, K.; Czajkowski, K.; Meczekalski, B. Premature ovarian insufficiency: The context of long-term effects. J. Endocrinol. Investig. 2016, 39, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamoda, H. The British Menopause Society and Women’s Health Concern recommendations on the management of women with premature ovarian insufficiency. Post Reprod. Heal. 2017, 23, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. J. Am. Med. Assoc. 2002, 288, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Boardman, H.M.P.; Hartley, L.; Eisinga, A.; Main, C.; Figuls, M.R.I.; Cosp, X.B.; Sanchez, R.G.; Knight, B. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst. Rev. 2015, 3, CD002229. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Walsh, J.M.E.; Greyber, E.; Salpeter, E.E. Brief report: Coronary heart disease events associated with hormone therapy in younger and older women. J. Gen. Intern. Med. 2006, 21, 363–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, J.E.; Aragaki, A.K.; Bassuk, S.S.; Chlebowski, R.T.; Anderson, G.L.; Rossouw, J.E.; Howard, B.V.; Thomson, C.A.; Stefanick, M.L.; Kaunitz, A.M.; et al. Menopausal Estrogen-Alone Therapy and Health Outcomes in Women With and Without Bilateral Oophorectomy: A Randomized Trial. Ann. Intern. Med. 2019, 171, 406. [Google Scholar] [CrossRef]
- Huang, A.J.; Sawaya, G.F.; Vittinghoff, E.; Lin, F.; Korenstein, D. Hot flushes, coronary heart disease, and hormone therapy in postmenopausal women. Menopause 2018, 25, 1286–1290. [Google Scholar] [CrossRef]
- Clarkson, T.B.; Meléndez, G.C.; Appt, S.E. Timing hypothesis for postmenopausal hormone therapy. Menopause J. North Am. Menopause Soc. 2013, 20, 342–353. [Google Scholar] [CrossRef]
- Sriprasert, I.; Hodis, H.N.; Karim, R.; Stanczyk, F.Z.; Shoupe, D.; Henderson, V.W.; Mack, W.J. Differential Effect of Plasma Estradiol on Subclinical Atherosclerosis Progression in Early vs Late Postmenopause. J. Clin. Endocrinol. Metab. 2019, 104, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Poornima, I.G.; Mackey, R.H.; Allison, M.A.; Manson, J.E.; Carr, J.J.; LaMonte, M.J.; Chang, Y.; Kuller, L.H.; Rossouw, J.; Ludlam, S.; et al. Coronary Artery Calcification (CAC) and Post-Trial Cardiovascular Events and Mortality Within the Women’s Health Initiative (WHI) Estrogen-Alone Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Mao, Y. Hormone treatments in congestive heart failure. J. Int. Med Res. 2018, 46, 2063–2081. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, K.; Abu Dabrh, A.M.; Benkhadra, K.; Al Nofal, A.; Carranza Leon, B.G.C.; Prokop, L.J.; Montori, V.M.; Faubion, S.S.; Murad, M.H. Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2015, 100, 4012–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezwada, P.; Shaikh, A.; Misra, D. The Effect of Transdermal Estrogen Patch Use on Cardiovascular Outcomes: A Systematic Review. J. Women’s Heal. 2017, 26, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Swica, Y.; Warren, M.P.; Manson, J.E.; Aragaki, A.K.; Bassuk, S.S.; Shimbo, D.; Kaunitz, A.; Rossouw, J.E.; Stefanick, M.L.; Womack, C.R. Effects of oral conjugated equine estrogens with or without medroxyprogesterone acetate on incident hypertension in the Women’s Health Initiative hormone therapy trials. Menopause 2018, 25, 753–761. [Google Scholar] [CrossRef]
- Cobin, R.H.; Goodman, N.F. American Association of Clinical Endoctinologists and American College of Endocrinology position statement on menopause–2017 update. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2017, 23, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Frisk, J. Managing hot flushes in men after prostate cancer—A systematic review. Maturitas 2010, 65, 15–22. [Google Scholar] [CrossRef]
- Freedland, S.J.; Eastham, J.; Shore, N. Androgen deprivation therapy and estrogen deficiency induced adverse effects in the treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2009, 12, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Bosco, C.; Bosnyak, Z.; Malmberg, A.; Adolfsson, J.; Keating, N.L.; Van Hemelrijck, M. Quantifying Observational Evidence for Risk of Fatal and Nonfatal Cardiovascular Disease Following Androgen Deprivation Therapy for Prostate Cancer: A Meta-analysis. Eur. Urol. 2015, 68, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Russell, N.; Hoermann, R.; Cheung, A.S.; Ching, M.; Zajac, J.D.; Handelsman, D.J.; Grossmann, M. Short-term effects of transdermal estradiol in men undergoing androgen deprivation therapy for prostate cancer: A randomized placebo-controlled trial. Eur. J. Endocrinol. 2018, 178, 565–576. [Google Scholar] [CrossRef]
- Gilbert, D.C.; Duong, T.; Kynaston, H.G.; Alhasso, A.A.; Cafferty, F.H.; Rosen, S.D.; Kanaga-Sundaram, S.; Dixit, S.; Laniado, M.; Madaan, S.; et al. Quality-of-life outcomes from the Prostate Adenocarcinoma: TransCutaneous Hormones (PATCH) trial evaluating luteinising hormone-releasing hormone agonists versus transdermal oestradiol for androgen suppression in advanced prostate cancer. BJU Int. 2016, 119, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Rutqvist, L.E.; Mattsson, A. The Stockholm Breast Cancer Study Group. Cardiac and Thromboembolic Morbidity among Postmenopausal Women with Early Stage Breast Cancer in a Randomized Trial of Adjuvant Tamoxifen. J. Natl. Cancer Inst. 1993, 85, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, S.; Shi, Y.; Smith, E.; Nattinger, A.B.; Laud, P.; Neuner, J. Are aromatase inhibitors associated with higher myocardial infarction risk in breast cancer patients? A Medicare population-based study. Clin. Cardiol. 2018, 42, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosrow-Khavar, F.; Bouganim, N.; Filion, K.B.; Suissa, S.; Azoulay, L. Cardiotoxicity of Use of Sequential Aromatase Inhibitors in Women With Breast Cancer. Am. J. Epidemiol. 2020, 189, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Qadir, H.; Amir, E.; Fischer, H.D.; Fu, L.; Austin, P.C.; Harvey, P.J.; Rochon, P.A.; Lee, D.S.; Anderson, G.M. The risk of myocardial infarction with aromatase inhibitors relative to tamoxifen in post-menopausal women with early stage breast cancer. Eur. J. Cancer 2016, 68, 11–21. [Google Scholar] [CrossRef]
- Walsh, B.W.; Kuller, L.H.; Wild, R.A.; Paul, S.; Farmer, M.; Lawrence, J.B.; Shah, A.S.; Anderson, P.W. Effects of raloxifene on serum lipids and coagulation factors in healthy postmenopausal women. JAMA 1998, 279, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Walsh, B.W.; Paul, S.; Wild, R.A.; Dean, R.A.; Tracy, R.P.; Cox, D.A.; Anderson, P.W. The Effects of Hormone Replacement Therapy and Raloxifene on C-Reactive Protein and Homocysteine in Healthy Postmenopausal Women: A Randomized, Controlled Trial1. J. Clin. Endocrinol. Metab. 2000, 85, 214–218. [Google Scholar] [CrossRef]
- Saitta, A.; Altavilla, D.; Cucinotta, D.; Morabito, N.; Frisina, N.; Corrado, F.; D’Anna, R.; Lasco, A.; Squadrito, G.; Gaudio, A.; et al. Randomized, Double-Blind, Placebo-Controlled Study on Effects of Raloxifene and Hormone Replacement Therapy on Plasma NO Concentrations, Endothelin-1 Levels, and Endothelium-Dependent Vasodilation in Postmenopausal Women. Arter. Thromb. Vasc. Biol. 2001, 21, 1512–1519. [Google Scholar] [CrossRef]
- Barrett-Connor, E.; Grady, D.; Sashegyi, A.; Anderson, P.W.; Cox, D.A.; Hoszowski, K.; Rautaharju, P.; Harper, K.D.; for the MORE Investigators. Raloxifene and Cardiovascular Events in Osteoporotic Postmenopausal Women. JAMA 2002, 287, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Taneja, S.S.; Smith, M.R.; Dalton, J.T.; Raghow, S.; Barnette, G.; Steiner, M.; A Veverka, K. Toremifene—A promising therapy for the prevention of prostate cancer and complications of androgen deprivation therapy. Expert Opin. Investig. Drugs 2006, 15, 293–305. [Google Scholar] [CrossRef]
- Sathyapalan, T.; Aye, M.; Rigby, A.S.; Thatcher, N.J.; Dargham, S.; Kilpatrick, E.S.; Atkin, S.L. Soy isoflavones improve cardiovascular disease risk markers in women during the early menopause. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 691–697. [Google Scholar] [CrossRef]
- Messina, M. Soy and Health Update: Evaluation of the Clinical and Epidemiologic Literature. Nutrients 2016, 8, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Nudy, M.; Aragaki, A.K.; Robbins, J.A.; Manson, J.E.; Stefanick, M.L.; O’Sullivan, D.M.; Shikany, J.M.; Leblanc, E.S.; Kelsey, A.M.; et al. Women’s Health Initiative clinical trials. Menopause 2019, 26, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Shearman, A.M.; Cupples, L.A.; Demissie, S.; Peter, I.; Schmid, C.H.; Karas, R.H.; Mendelsohn, M.E.; Housman, D.E.; Levy, D. Association Between Estrogen Receptor α Gene Variation and Cardiovascular Disease. J. Am. Med. Assoc. 2003, 290, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Shearman, A.M.; Cooper, J.A.; Kotwinski, P.; Miller, G.J.; Humphries, S.E.; Ardlie, K.G.; Jordan, B.; Irenze, K.; Lunetta, K.L.; Schuit, S.C.E.; et al. Estrogen Receptor α Gene Variation Is Associated With Risk of Myocardial Infarction in More Than Seven Thousand Men From Five Cohorts. Circ. Res. 2006, 98, 590–592. [Google Scholar] [CrossRef] [Green Version]
- Schuit, S.C.E.; Oei, H.-H.S.; Witteman, J.C.M.; Van Kessel, C.H.G.; Van Meurs, J.B.J.; Nijhuis, R.L.; Van Leeuwen, J.P.T.M.; De Jong, F.H.; Zillikens, M.C.; Hofman, A.; et al. Estrogen Receptor α Gene Polymorphisms and Risk of Myocardial Infarction. JAMA 2004, 291, 2969–2977. [Google Scholar] [CrossRef] [Green Version]
- Henttonen, A.T.; Kortelainen, M.; Kunnas, T.A.; Nikkari, S.T. Estrogen receptor-1 genotype is related to coronary intima thickness in young to middle-aged women. Scand. J. Clin. Lab. Investig. 2007, 67, 380–386. [Google Scholar] [CrossRef]
- Roszkowska-Gancarz, M.; Kuryłowicz, A.; Polosak, J.; Ambroziak, M.; Puzianowska-Kuznicka, M. The −351A/G polymorphism of ESR1 is associated with risk of myocardial infarction but not with extreme longevity. Clin. Chim. Acta 2010, 411, 1883–1887. [Google Scholar] [CrossRef]
- Domingues-Montanari, S.; Subirana, I.; Tomás, M.; Marrugat, J.; Sentí, M. Association between ESR2 Genetic Variants and Risk of Myocardial Infarction. Clin. Chem. 2008, 54, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Ambroziak, M.; Kuryłowicz, A.; Roszkowska-Gancarz, M.; Budaj, A. ESR2 gene G1730A variant is associated with triglycerides level and myocardial infarction in young men but not in women. Gene 2018, 677, 83–88. [Google Scholar] [CrossRef]
- Kuźbicka, K.; Rachoń, D.; Woziwodzka, A.; Rybicka, M.; Bielawski, K.P. Associations of ESR1 and ESR2 gene polymorphisms with metabolic syndrome and its components in postmenopausal women. Maturitas 2018, 115, 97–102. [Google Scholar] [CrossRef]
- Manosroi, W.; Tan, J.W.; Rariy, C.M.; Sun, B.; Goodarzi, M.O.; Saxena, A.R.; Williams, J.S.; Pojoga, L.H.; Lasky-Su, J.; Cui, J.; et al. The Association of Estrogen Receptor-β Gene Variation with Salt-Sensitive Blood Pressure. J. Clin. Endocrinol. Metab. 2017, 102, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liang, G.; Pei, Y.; Ye, W.; Liang, A.; Su, P. Genomic polymorphisms of G-Protein Estrogen Receptor 1 are associated with severity of adolescent idiopathic scoliosis. Int. Orthop. 2011, 36, 671–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, N.; Paul-Bellon, R.; Camparo, P.; Michiels, J.-F.; Chevallier, D.; Fénichel, P. Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma. Int. J. Mol. Sci. 2014, 15, 1574–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasap, B.; Turhan, N.O.; Edgunlu, T.; Duran, M.; Akbaba, E.; Oner, G. G-protein-coupled estrogen receptor-30 gene polymorphisms are associated with uterine leiomyoma risk. Bosn. J. Basic Med. Sci. 2015, 16, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Fredette, N.C.; Malik, E.; Mukhtar, M.L.; Prossnitz, E.R.; Terada, N. A hypertension patient-derived iPSC model demonstrates a role for G protein-coupled estrogen receptor in hypertension risk and development. Am. J. Physiol. Cell Physiol. 2020, 319, C825–C838. [Google Scholar] [CrossRef]


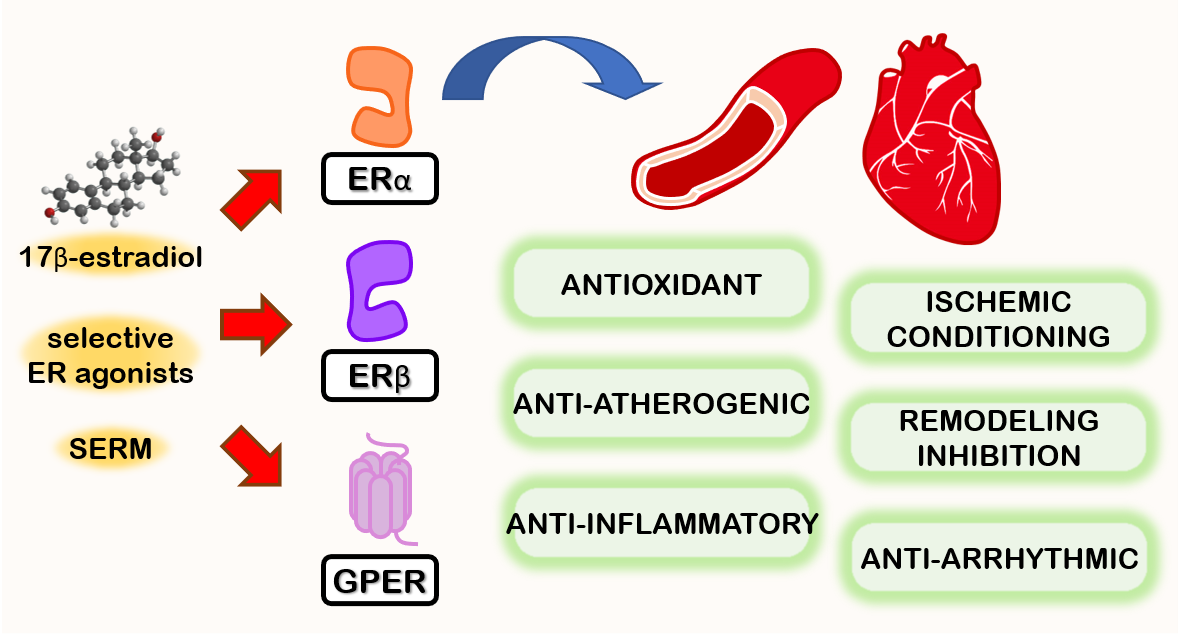{kind=link}
{kind=link}
| Agonist | Receptor | Model | Effect | Mechanism | Ref. |
|---|---|---|---|---|---|
| E2 | Unknown | acute and chronic OVX-MI—Rats | ↑ infarcted area ↓ ventricular wall tension ↓ ventricular dilatation ↑ survival | ↑ endothelin B receptor | [41] |
| E2 | Unknown | I/R—Male dogs | ↓ infarcted area ↓ arrhythmias | opens K+ channels ↑ NO production | [42] |
| E2 | Unknown | OVX-MI—Mice | ↓ cardiomyocyte apoptosis ↓ infarcted area | PI3K/Akt activation | [29,43] |
| E2 | Unknown | Staurosporine-induced apoptosis—Rat cardiomyocytes | ↓ cardiomyocyte apoptosis | NF-κB inhibition | [44,45] |
| E2 | Unknown | AngII-induced hypertrophy—Rat cardiomyocytes | ↓ hypertrophy and remodeling gene transcription | calcineurin inhibition (via PI3K) | [46] |
| E2 | ERβ (ERα if under stress) | OVX + high-fat diet—Mice | Improves cardiovascular function | ↑ SOD2 (heart and aorta) | [47] |
| E2 | ERα > ERβ | acute and chronic OVX-MI—Mice | ↓ injured myocardium ↑ angiogenesis | activation of bone marrow-derived EPC | [48,49,50] |
| E2, E2-BSA, PPT | ERα | OVX-MI—Rats | ↓ cardiac fibrosis | ↓ cofilin phosphorylation (↓ RhoA/ROCK activity) | [51] |
| E2, DPN | ERβ | OVX + I/R—Mice | ↓ I/R myocardial injury | ↑ cardioprotective genes (COX2, GADD45β, IL-6, heat shock proteins, PFKFB) | [52] |
| E2, DPN | ERβ | OVX + I/R—Mice | Improves cardiac function ↓ infarcted area | ↑ cardiac protein SNO | [53] |
| G1 | GPER | OVX + atherogenic—Mice | ↓ atherosclerosis ↓ vascular inflammation | ↓ vessel inflammation ↑ NO production | [23] |
| E2, βLGND, DPN | ERβ | AngII-induced fibrosis—Rat cardiac fibroblasts | ↓ extracellular matrix production | ↓ ROCK activity ↓ TGF-β production (via AMPK and PKA) | [54] |
| E2, WAY-169916 | ERα/ERβ | OVX + I/R—Rabbit | ↓ infarcted area ↓ neutrophil accumulation | NF-κB inhibition | [55] |
| E2, G-1 | GPER | OVX + Ca2+ supplementation—Rats | ↓ total cholesterol | hepatic TRPC1 inhibition, ↓ intracellular Ca2+ | [56] |
| G-1 | GPER | I/R—Male mice, female and male rats | Improves cardiac function ↓ infarction area | ↓ oxidative stress ↓ ATP depletion ↓ mPTP opening (via PI3K/Akt) | [20,22,57] |
| E2, PPT, G-1 | ERα/GPER | OVX-MI—Rats | ↓ infarction size ↓ myocardial fibrosis | ↑ eNOS activity (via PI3K/Akt) | [58] |
| G-1 | GPER | OVX-MI—Mice | Improved cardiac function ↓ apoptosis ↓ myocardial inflammation | ↓ TNF-α ↑ IL-10 (via PI3K/Akt) | [59] |
| Strain | Model | Phenotype | Mechanism | Ref. |
|---|---|---|---|---|
| Targeted endothelial ErβOE—Male rats | I/R | ↓ oxidative burst ↓ vascular injury | ↑ ERRα, SOD2, eNOS expression ↑ NO/ROS ratio ↑ mitochondrial function | [60] |
| Targeted cardiac ErβOE—Female mice | MI | ↓ ventricle dilatation ↓ fibrosis | ↓ fibrotic gene program expression | [61] |
| ERβKO—Female and male mice | I/R | ↑ cardiac injury after isoproterenol (females) | ↓ recovery of intracellular ATP level ↑ intracellular acidification ↓ eNOS, lipogenic enzymes | [62] |
| ERβKO—Mice | OVX + AngII | ↓ E2 anti-hypetrophy and anti-fibrosis effects | ↓ PI3K/MCIP1 inhibition of hypertrophy and fibrosis gene programs | [26,63] |
| ErβKO—Mice | OVX + I/R | ↑ cardiac damage | ↓ PI3K/Akt pathway ↑ apoptotic signaling | [64] |
| ErβKO—Female mice | MI | ↑ ventricular repolarization time ↓ ventricular premature beats | ↓ mRNA levels of Kv4.3 | [65] |
| GPERKO—Mice | OVX and atherogenic diet | ↑ atherosclerosis progression | ↑ vascular inflammation ↓ vascular basal NO production | [23] |
| GPERKO—Mice | OVX and atherogenic diet | ↓ anti-atherogenic action of E2 ↓ coronary vasodilatation | ↑ endothelium-dependent contractions to vasoconstrictor prostanoids | [66] |
| GPERKO—Male mice | I/R | ↓ E2-improvement in cardiac function ↑ infarcted area ↑mitochondrial Ca2+ overload | ↓ activation of MEK/ERK pathway GSK-3β | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, J.S.; Montagnoli, T.L.; Rocha, B.S.; Tacco, M.L.C.A.; Marinho, S.C.P.; Zapata-Sudo, G. Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 525. https://doi.org/10.3390/ijms22020525
da Silva JS, Montagnoli TL, Rocha BS, Tacco MLCA, Marinho SCP, Zapata-Sudo G. Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. International Journal of Molecular Sciences. 2021; 22(2):525. https://doi.org/10.3390/ijms22020525
Chicago/Turabian Styleda Silva, Jaqueline S., Tadeu L. Montagnoli, Bruna S. Rocha, Matheus L. C. A. Tacco, Sophia C. P. Marinho, and Gisele Zapata-Sudo. 2021. "Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction" International Journal of Molecular Sciences 22, no. 2: 525. https://doi.org/10.3390/ijms22020525
APA Styleda Silva, J. S., Montagnoli, T. L., Rocha, B. S., Tacco, M. L. C. A., Marinho, S. C. P., & Zapata-Sudo, G. (2021). Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. International Journal of Molecular Sciences, 22(2), 525. https://doi.org/10.3390/ijms22020525