Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases



Abstract
:1. Introduction
2. Results
2.1. Mitochondrial Replacement Procedures and Variables Measured
- Polar Body Transfer:
- (1)
- Potential embryo development of the reconstituted oocyte/zygote: this variable informs about the survival rate of the reconstituted oocyte/zygote after its micromanipulation, and its potential to progress to different development stages (morula, blastocyst, or even birth in animals) [19,27,28]. In humans, generally the development reached morula or blastocyst stages and fetuses survived only to mid-gestation [1,35]. However, another work [34] reported the birth of a healthy boy from a mother carrying mutated mtDNA. Given the ethical and legal restrictions on human embryo research, many authors decided to deeply study the potential development of embryonic stem cells (ESCs) from blastocysts [29,30,32,33,36,37,39,40], although these cells may not be representative of normal embryo cells [1].
- (2)
- Mitochondrial DNA carry-over: mitochondrial carry-over is defined as the mtDNA that is inadvertently transferred with the nDNA, since, to date, 100% mitochondrial exclusion has not been possible. This carry-over is expressed as heteroplasmy percentage: the ratio of maternal mtDNA with respect to the donor ovum mtDNA.
2.2. Germinal Vesicle Transfer (GVT)
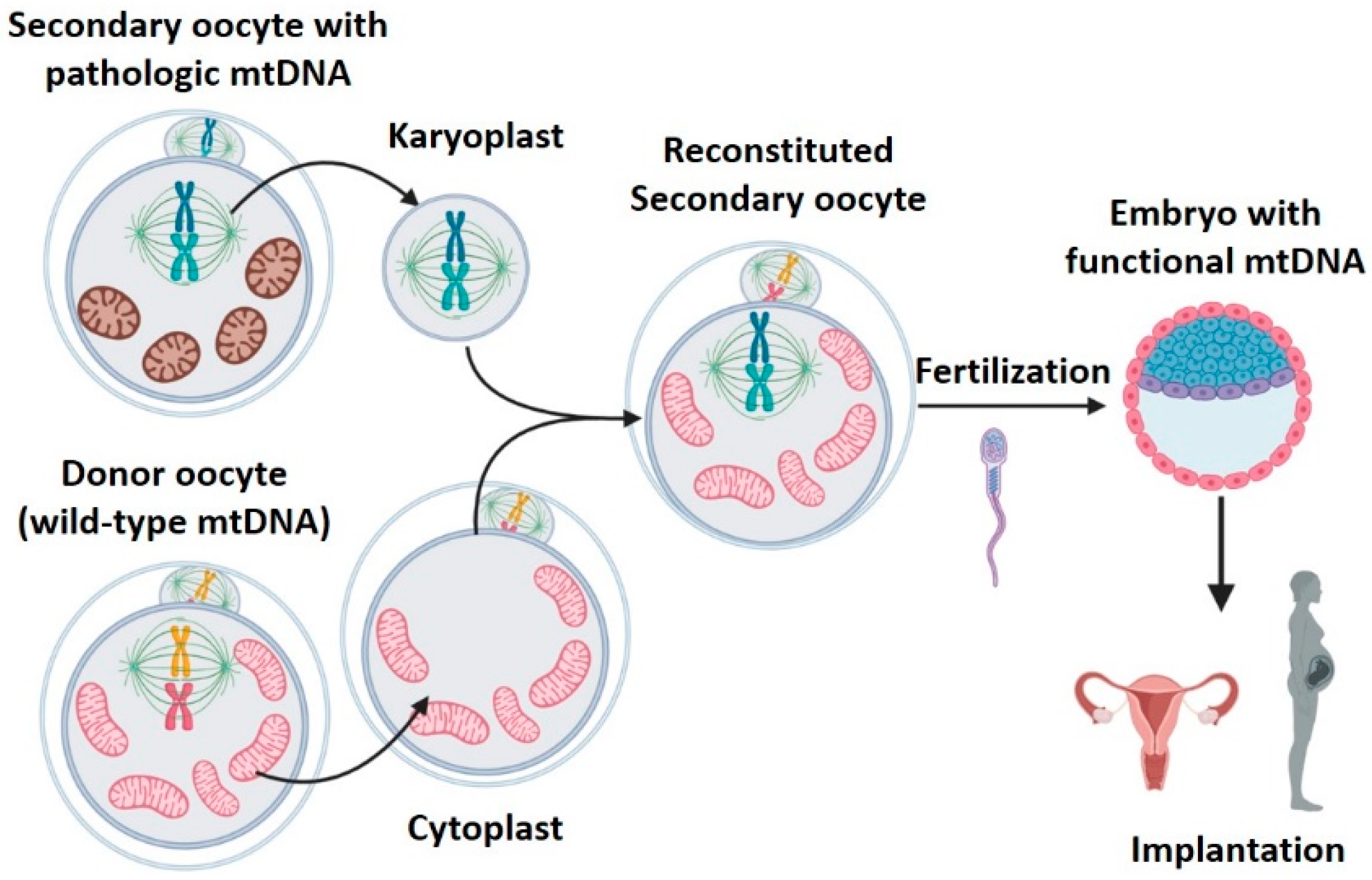
2.3. Meiotic Spindle Transfer (MST)
- 1.
- High efficacy: reconstituted oocytes presented rates of development to blastocyst equivalent to those obtained in controls [19]. Furthermore, the four macaques born remained healthy in both physical (weight, height, etc.) and analytical explorations during 3 years follow-up [30]. MST was compatible with normal embryo growth, and was able to generate healthy individuals.
- 2.
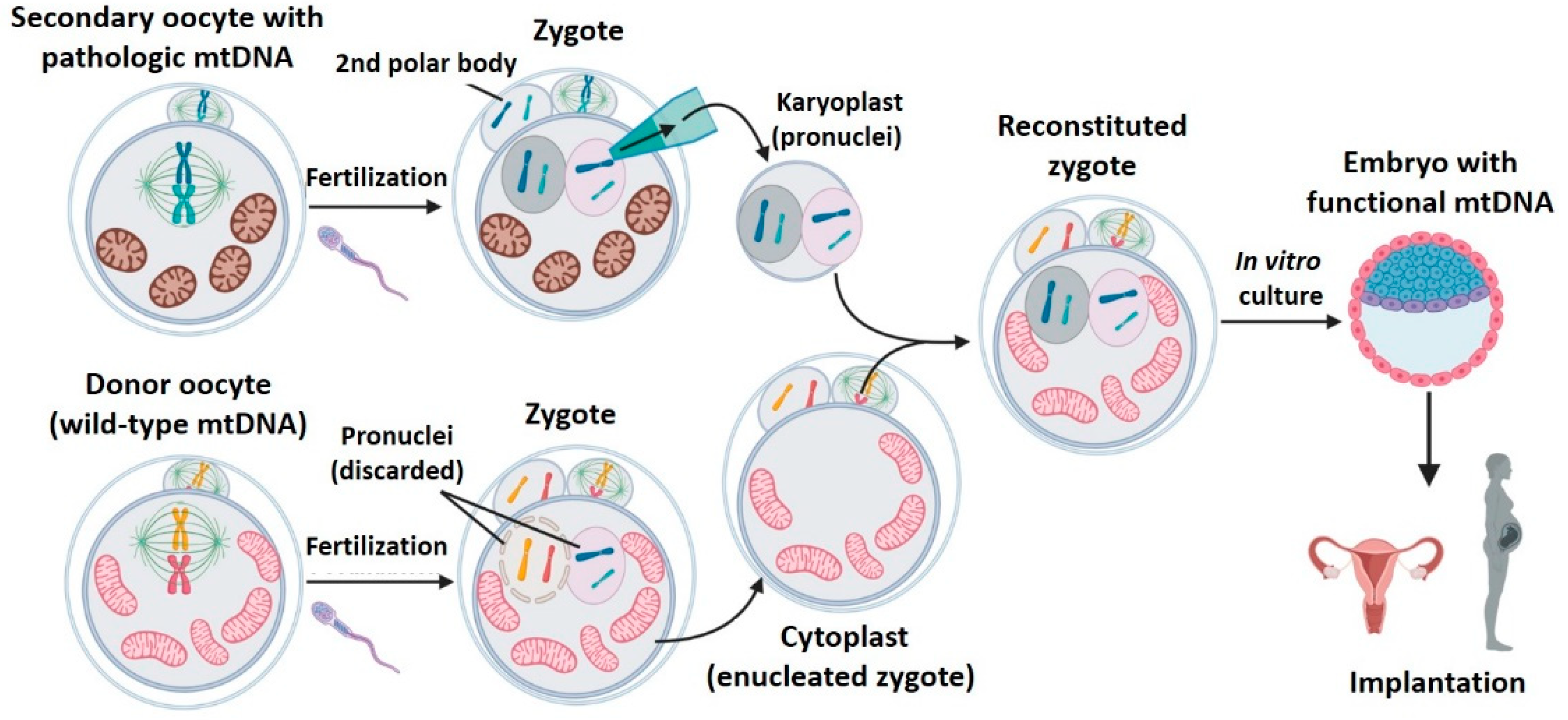
2.4. Pronuclear Transfer (PNT)
2.5. Polar Bodies Transfer (PB1T/PB2T)
- First polar body transfer (PB1T): at the end of meiosis I, a secondary oocyte (containing most of the primary oocyte cytoplasm) and the first polar body, which is extruded to the cell periphery, are obtained. The secondary oocyte and first polar body genomes are practically identical [31]. That is, formed by 23 chromosomes (n) with two chromatids each (2c). Unlike the metaphase spindle of the secondary oocyte, the first polar body spindle is clearly delimited by a surrounding plasmatic membrane, with a low amount of cytoplasm. The first polar body transfer (PB1T) consists of transferring the first polar body from a patient’s oocyte in metaphase II to a donor oocyte, also in metaphase II, without meiosis spindle (previously removed) [31,38,39,40] (Figure 6). The resulting oocyte will be fertilized and implanted. This technique is very similar to MST, but the meiotic spindle in the recipient enucleated oocyte is replaced by the first polar body.
- Second polar body transfer (PB2T): when the secondary oocyte is fertilized, meiosis II ends to form a female pronuclei whose chromosomes have one unique chromatid (from (n, 2c) to (n, c)). The second meiotic division also generates the second polar body (PB2) whose genetic material is similar to that of the zygote’s female pronuclei (n, c). As occurs with PB1, PB2 is well delimited in the zygote’s periphery and has little cytoplasm. PB2T consists in transferring the PB2 from a patient’s zygote to a donor zygote whose female pronuclei has been previously removed (since it contains donor nDNA) [31,38,40] (Figure 7). As in PNT, both PB1T and PB2T require zygote manipulation. First polar body transfer (PB1T) consists in transferring the first polar body (PB1) from the secondary maternal oocyte to a donor oocyte previously enucleated (without meiotic spindle). Thus, this technique is similar to mother spindle transfer. In contrast, second polar body transfer (PB2T) implies isolating the second polar body (PB2) of a maternal zygote (PB2) and substituting the female pronuclei of a donor zygote. PB2T is similar to PNT since both of them use zygotes.
2.6. Benefits and Risks of Mitochondrial Procedure
2.6.1. Comparison of Mitochondrial Replacement Techniques (MRTs)
2.6.2. Risks Associated to Mitochondrial Replacement Therapies
- Risks derived from micromanipulation
- Mitochondrial carry-over
- Mito-nuclear incompatibility
- (a)
- Sendai virus extract (HVJ-E): it is employed to fuse the karyoplast with the cytoplast. Although this extract is purified and only contains envelope proteins, it could contain traces of viral genome RNA with the potential to integrate in the embryo genome [19]. The possible repercussions of this potential scenario are unknown. The rhesus macaques that were born employing spindle transfer did not have the presence of viral genetic material within their genome [19]. However, there remains some reticence to using viral proteins as fusogens in a clinical setting [38]. In this sense, Zhang et al. [34] chose the electrofusion procedure for their spindle transfer intervention that led to the first healthy human male birth, aiming to avoid introducing foreign proteins [34]. More information about its safety will be required prior to its clinical translation.
- (b)
- Cytoskeletal disruptors (cytochalasin B, nocodazole): during nuclear transfer procedures, the oocytes are incubated with cytoskeleton disruptors to facilitate their manipulation [19,20]. Although they are also used frequently in techniques of in vitro fertilization, their possible deleterious effects have not been evaluated properly [37]. Only Wu et al. [37,40] modified the transfer procedures to avoid the administration of disruptors, obtaining positive results.
- (c)
- Nuclear genome damage: instrumental stress over oocyte genetic material could cause chromosomal damage and increase embryonic aneuploidies [38]. This could be more probable when transferring the meiotic spindle, because it is not protected by a membrane, however no information about this was found.
2.7. Ethical and Legal Aspects of Mitochondrial Replacement
- Do MRTs constitute a germinal gene therapy?
- Do the individuals born by MRTs have three parents (2 mothers and 1 father)?
- Are MRTs ethically justifiable?
- What is the legal status of MRTs? What are their indications?
2.7.1. Should MRTs Be Considered a Germinal Gene Therapy?
2.7.2. Are They Really Children of 3 Parents?
2.7.3. Are MRTs Ethically Justifiable?
2.7.4. Which Is the Legal Status of MRTs? What Are Their Indications?
3. Conclusions
- Germinal vesicle transfer is not efficient in human oocytes.
- Spindle transfer has accumulated a wide experience, including human gestations.
- Pronucleus transfer is an efficient technique, extensively described in the literature, but presents ethical concerns as it requires zygote destruction.
- First polar body transfer is a promising strategy with excellent preclinical results, but further studies will be required prior to its application.
- Second polar body transfer presents technical difficulties in human zygotes.
- MRTs reduce the risk of vertical transmission of mitochondrial diseases, but they do not completely prevent it. For this reason, it is recommended:
- To minimize the mitochondrial drag by choosing those procedures with the lowest intrinsic carry-over (polar bodies).
- To use oocytes from donors with a compatible haplogroup.
- Selecting male embryos.
- Long-term follow-up of animals and humans born by MRTs.
- It is mandatory that governments legislate about the use of MRTs, especially to define those clinical applications in which the risks surpass the potential benefits.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenfield, A.; Braude, P.; Flinter, F.; Lovell-Badge, R.; Ogilvie, C.; Perry, A.C.F. Assisted reproductive technologies to prevent human mitochondrial disease transmission. Nat. Biotechnol. 2017, 35, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.F. Mitochondrial Genetics. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 247–255. [Google Scholar] [CrossRef]
- Tzameli, l. The evolving role of mitochondria in metabolism. Trends Endocrinol. Metab. 2012, 23, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Med. 2013, 3, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.M.; Sondheimer, N. Heteroplasmy Shifting as Therapy for Mitochondrial Disorders. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; Volume 1158, pp. 257–267. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craven, L.; Murphy, J.; Turnbull, D.M.; Taylor, R.W.; Gorman, G.S.; McFarland, R. Scientific and Ethical lssues in Mitochondrial Donation. New Bioeth. 2018, 24, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.W.; Taylor, G.A.; Durham, S.E.; Turnbull, D.M. The determination of complete human mitochondrial DNA sequences in single cells: Implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res. 2001, 29, e74. [Google Scholar] [CrossRef]
- Reinecke, F.; Smeitink, J.A.; Van Der Westhuizen, F.H. OXPHOS gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta Mol. Basis Oís. 2009, 1792, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Urbani, A.; De Canio, M.; Palmieri, F.; Sechi, S.; Bini, L.; Castagnola, M.; Fasano, M.; Modesti, A.; Roncada, P.; Timperio, A.M.; et al. The mitochondrial ltalian Human Proteome Project initiative (mt-HPP). Mol. Biosyst. 2013, 9, 1984–1992. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Distelmaier, F.; Smeitink, J.A.M.; Willems, P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, I.; Shutt, T.E. Advances towards Therapeutic Approaches for mtDNA Disease. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; Volume 1158, pp. 217–246. [Google Scholar] [CrossRef]
- Smeets, H.J.M. Preventing the transmission of mitochondrial DNA disorders: Selecting the good guys or kicking out the bad guys. Reprod. Biomed. Online 2013, 27, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Pompei, M.; Pompei, F. Overcoming bioethical, legal, and hereditary barriers to mitochondrial replacement therapy in the USA. J. Assist. Reprod. Genet. 2019, 36, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Craven, L.; Elson, J.L.; Irving, L.; Tuppen, H.A.L.; Lister, L.M.; Greggains, G.D.; Byerley, S.; Murdoch, A.P.; Herbert, M.; Turnbull, D.M. Mitochondrial DNA disease: New options for prevention. Hum. Mol. Genet. 2011, 20, R168–R174. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Grady, J.P.; Turnbull, D.M. Mitochondrial donation - How many women could benefit? N. Engl. J. Med. 2015, 372, 885–887. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Tatay, L.; Hernández-Andreu, J.; Aznar, J. Mitochondrial Modification Techniques and Ethical lssues. J. Clin. Med. 2017, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Kuno, T.; Yaegashi, N. Mitochondrial replacement therapy and assisted reproductive technology: A paradigm shift toward treatment of genetic diseases in gametes or in early embryos. Reprod. Med. Biol. 2018, 17, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klitzman, R.; Toynbee, M.; Sauer, M.V. Controversies concerning mitochondrial replacement therapy. Fertil. Steril. 2015, 103, 344–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios-González, C. Mexico and mitochondrial replacement techniques: What a mess. Br. Med. Bull. 2018, 128, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, K.; Kellam, L.D.; Lee, Y.S.; Liang, C.-G.; Han, Z.; Mtango, N.R.; Latham, K.E. Effects of Ooplasm Manipulation on ONA Methylation and Growth of Progeny in Mice. Biol. Reprod. 2009, 80, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Neupane, J.; Vandewoestyne, M.; Ghimire, S.; Lu, Y.; Qian, C.; Van Coster, R.; Gerris, J.; DeRoo, T.; Deforce, D.; De Sutter, P.; et al. Assessment of nuclear transfer techniques to prevent the transmission of heritable mitochondrial disorders without compromising embryonic development competence in mice. Mitochondrion 2014, 18, 27–33. [Google Scholar] [CrossRef]
- Paull, D.; Emmanuele, V.; Weiss, K.A.; Treff, N.; Stewart, L.; Hua, H.; Zimmer, M.; Kahler, D.J.; Goland, R.S.; Noggle, S.A.; et al. Nuclear genome transfer in human oocytes eliminates mitochondrial ONA variants. Nature 2013, 493, 632–637. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.-S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013, 493, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Sha, H.; Ji, D.; Zhang, H.L.; Chen, D.; Cao, Y.; Zhu, J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 2014, 157, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Emmanuele, V.; Sanchez-Quintero, M.J.; Sun, B.; Lallos, G.; Paull, D.; Zimmer, M.; Pagett, S.; Prosser, R.W.; Sauer, M.V.; et al. Genetic Orift Can Compromise Mitochondrial Replacement by Nuclear Transfer in Human Oocytes. Cell Stem Cell 2016, 18, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Luo, S.; Lu, Z.; Chávez-Badiola, A.; Liu, Z.; Yang, M.; Merhi, Z.; Silber, S.J.; Munné, S.; et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod. Biomed. Online 2017, 34, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhuang, G.; Zeng, Y.; Grifo, J.; Acosta, C.; Shu, Y.; Liu, H. Pregnancy derived from human zygote pronuclear transfer in a patient who had arrested embryos after IVF. Reprod. Biomed. Online 2016, 33, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.E.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Chen, T.; Huang, S.; Zhong, C.; Yan, J.; Zhang, X.; Li, J.; Gao, Y.; Zhao, H.; Chen, Z.-J. Mitochondrial replacement by pre-pronuclear transfer in human embryos. Cell Res. 2017, 27, 834–837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.P.; Lu, C.F.; Gong, F.; Xie, P.Y.; Hu, L.; Zhang, S.J.; Lu, G.X.; Lin, G. Polar body transfer restares the developmental potential of oocytes to blastocyst stage in a case of repeated embryo fragmentation. J. Assist. Reprod. Genet. 2017, 34, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; O’Neil, R.C.; Gutierrez, N.M.; Hariharan, M.; Zhang, Z.Z.; He, Y.; Cinnioglu, C.; Kayali, R.; Kang, E.; Lee, Y.; et al. Functional Human Oocytes Generated by Transfer of Polar Body Genomes. Cell Stem Cell 2017, 20, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Zhong, C.; Chen, T.; Zhang, X.; Tao, W.; Zhang, J.; Li, H.; Zhao, H.; Li, J.; Chen, Z.-J. Polar bodies are efficient donors for reconstruction of human embryos for potential mitochondrial replacement therapy. Cell Res. 2017, 27, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Sloan, O.B.; Fields, P.O.; Havird, J.C. Mitonuclear linkage disequilibrium in human populations. Proc. R. Soc. B Boil. Sci. 2015, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, J.; Solter, D. Nuclear transplantation in the mouse embryo by microsurgery and cell fusion. Science 1983, 220, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kono, T.; Nakada, K.; Ishikawa, K.; Inoue, S.-I.; Yonekawa, H.; Hayashi, J.-I. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc. Natl. Acad. Sci. USA 2005, 102, 16765–16770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, N.; Prendergast, D.; Oehlert, J.W.; Shaw, G.M.; Stevenson, D.K.; Rappaport, N.; Sirota, M.; Tishkoff, S.A.; Sondheimer, N. Divergent Patterns of Mitochondrial and Nuclear Ancestry Are Associated with the Risk for Preterm Birth. J. Pediatr. 2018, 194, 40–46.e4. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.A.; Makova, K.D. lnvestigating mitonuclear interactions in human admixed populations. Nat. Ecol. Evol. 2019, 3, 213–222. [Google Scholar] [CrossRef]
- Klucnika, A.; Ma, H. A battle for transmission: The cooperative and selfish animal mitochondrial genomes. Open Biol. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havird, J.C.; Hall, M.O.; Oowling, O.K. The evolution of sex: A new hypothesis based on mitochondrial mutational erosion. BioEssays 2015, 37, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobler, R.; Dowling, D.K.; Morrow, E.H.; Reinhardt, K. A systematic review and meta-analysis reveals pervasive effects of germline mitochondrial replacement on components of health. Hum. Reprod. Update 2018, 24, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Rishishwar, L.; Jordan, I.K. lmplications of human evolution and admixture for mitochondrial replacement therapy. BMC Genom. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre-Walker, A. Mitochondrial replacement therapy: Are mito-nuclear interactions likely to be a problem? Genetics 2017, 205, 1365–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Graven, L.; Mitalipov, S.; Stewart, J.B.; Herbert, M.; Turnbull, O.M. The Challenges of Mitochondrial Replacement. PLoS Genet. 2014, 10, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T. Potential impact of human mitochondrial replacement on global policy regarding germline gene modification. Reprod. Biomed. Online 2014, 29, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Baylis, F. The ethics of creating children with three genetic parents. Reprod. Biomed Online 2013, 26, 531–534. [Google Scholar] [CrossRef] [Green Version]
- Claiborne, A.B.; English, R.A.; Kahn, J.P. Finding an ethical path forward for mitochondrial replacement. Science 2016, 351, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Newson, A.J.; Wilkinson, S.; Wrigley, A. Ethical and legal issues in mitochondrial transfer. EMBO Mol. Med. 2016, 8, 589–591. [Google Scholar] [CrossRef] [Green Version]
- Adashi, E.Y.; Cohen, I.G. Preventing Mitochondrial Diseases: Embryo-Sparing Donor-lndependent Options. Trends Mol. Med. 2018, 24, 449–457. [Google Scholar] [CrossRef]
- Dimond, R. Social and ethical issues in mitochondrial donation. Br. Med. Bull. 2015, 115, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, M.; Turnbull, O. Mitochondrial replacement to prevent the transmission of mitochondrial ONA disease. EMBO Rep. 2015, 16, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Engelstad, K.; Sklerov, M.; Kriger, J.; Sanford, A.; Grier, J.; Ash, D.; Egli, D.; DiMauro, S.; Thompson, J.L.; Sauer, M.V.; et al. Attitudes toward prevention of mtDNA-related diseases through oocyte mitochondrial replacement therapy. Hum. Reprod. 2016, 31, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Bredenoord, A.L.; Appleby, J.B. Mitochondrial Replacement Techniques: Remaining Ethical Challenges. Cell Stem Cell 2017, 21, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Hibino, Y. Mitochondrial manipulation in fertility clinics: Regulation and responsibility. Reprod. Biomed. Soc. Online 2018, 5, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zukin, V.; Alexandrova, S.; Mazur, P.; Mykytenko, D.; Liu, H.; Dyachenko, L. Clinical trial for the treatment of infertility due to poor oocyte quality. ISRCTN 2019, 13358803. [Google Scholar] [CrossRef]
- Kostaras, K.; Costa-Borges, N.; Psathas, P.; Calderón, G.; Nikitos, E. Spindle transfer for the treatment of infertility problems associated to poor egg quality: A pilot trial. ISRCTN 2018, 11455145. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Procedure/ Characteristics | GVT | MST | PNT | PB1T | PB2T |
|---|---|---|---|---|---|
| Origin | Primary oocyte | Secondary oocyte | Zygote | Secondary oocyte | Zygote |
| Ploidy | Diploid (2n) | Haploid (n) | Diploid (2 × n, mas. and fem.) | Haploid (n) | Haploid (n) |
| Chromatids | 4C | 2C | 2 × C (masc. and fem.) | 2C | C |
| Surrounding membrane | + | - | + | + | + |
| Ref. | Transfer Method | Blastocyst Development | Births Rate | Carry-Over | Notes |
|---|---|---|---|---|---|
| Cheng et al., 2009 [31] | Electrofusion (EF) | Not indicated | 7.7% | No tested | Lower post-natal growth |
| Neupane et al., 2014 [36] | HVJ-E | Inter-strain: 0% (0/14) Intra-strain: 0% (0/8) | N/D | Reconstructed oocytes: 0% (0/20) | MST and PNT were also compared |
| Ref. | Transfer Method | Blastocyst Development | Births Rate | Carry-Over | Notes |
|---|---|---|---|---|---|
| Tachibana et al., 2009 [19] | HVJ-E EF | HVJ-E: 61% (45/74) similar to control EF: 9% (1/11) lower than control | 27% (4/15) 17% Similar to control | ESCs and offspring: undetectable (<3%) | First animals born by MST Lower development to blastocyst due to premature oocyte activation |
| Cheng et al., 2009 [27] | EF | Not indicated | Inter-strain: 26% Intra-strain: 34% Similar to control | Not tested | GVT and PNT also performed |
| Neupane et al., 2014 [28] | HVJ-E | PTGN: 82.6% (19/23) similar to control ICSI: 0% (0/12) similar to control | Not tested | 0.29% ± 0.63% Undetectable in 17/24 | Carry-over of MST, GVT, and PNT were compared |
| Wang et al., 2014 [31] | HVJ-E | 85.7% (18/21) | 44.4% (8/18) | F1 tail: 5.5% ± 1.4% F2 fingers: 7.1% ± 6.8% | First publication to propose polar bodies transfer |
| Ref. | Fertil. Proc. | Transfer Method | Blastocyst Development | Births Rate | Carry-Over | Notes |
|---|---|---|---|---|---|---|
| Tachibana et al., 2013 [30] | ICSI | HVJ-E | 43% (19/44) lower than control | Not tested | Embryos: 0.5% ± 0.4% ESCs: 0.6% ± 0.9% | First MST in human oocytesAbnormal fertilization in 52% zygotes |
| Paull et al., 2013 [29] | PTGN | HVJ-E EF | 38.9 (7/18) 33% similar to control | Not tested | Embryos: 0.31% ± 0.27% ESCs: <0.5% * * except 1 line (P4-P14) 2.79% | Prevention of spindle activation by low-temperature electrofusion |
| Yamada et al., 2016 [32] | PTGN | Not indicated | 32% (cryogenized karyoplast + fresh cytoplast) | Not tested | Embryos: 0.2% ESCs: 0% in 7/8 cell lines (P6-P30) In 1/8 cell lines P0-1%; P36 = 53%; P59 = 1% | Complete reversion to biological mother mitochondrial haplotype in ESCs derived from SCNT |
| Kang et al., 2016 [33] | ICSI | HVJ-E | Healthy: 62.5% (20/32) Mutation carriers: 50% (6/12) | Not tested | Embryos: <1% ESCs: 15/18 <1% 3/18 100% | Ovum from pathological mtDNA carriers Improved abnormal fertilization |
| Zhang et al., 2017 [34] | ICSI | EF | 80% (4/5) | 100% (1/1) | Blastocyst: 5.10% ± 1.11% Urine: 2.36% Mouth: 5.59% Foreskin: 9.23% | First human birth by mitochondrial replacement technique |
| Ref. | Transfer Method | Blastocyst Development | Births Rate | Carry-Over | Notes |
|---|---|---|---|---|---|
| McGrath et al., 1983 [42] | EF | 96% (64/67) Similar to controls | 15% (10/64) Similar to controls | Not tested | First publication about PNT |
| Sato et al., 2005 [43] | EF | Not indicated | 28% (11/39) Similar to controls | F1 tail: 11% (6–21%) After 300 days: 23% (5–44%) | Murine model with mitochondrial pathology. Offspring free of disease |
| Cheng et al., 2009 [27] | EF | Not indicated | 31–62% | Not tested | Animals born were healthy and comparable to controls |
| Neupane et al., 2014 [28] | HVJ-E | 87.5% (14/16) Similar to non-manipulated controls | Not tested | Embryos: 0.29% ± 0.75% Undetectable in 21/25 | Minimum levels of carry-over. Equivalent to those of GVT and MST |
| Wang et al., 2014 [31] | HVJ-E | 81.3% (13/16) Similar to controls | 53% (7/13) Similar to controls | F1 tail: 23.7% ± 11.1% F2 fingers: 22.1% ± 18.7% | High rates of carry-over, probably due to manipulation problems |
| Ref. | Transfer Method | Blastocyst Development | Births Rate | Carry-Over | Notes |
|---|---|---|---|---|---|
| Craven et al., 2010 [20] | HVJ-E | 8.3% lower than control | Not tested | Embryos: 8.1% ± 7.6% Embryos (careful manipulation): 1.68% ± 1.81% | Employed abnormally fertilized embryos |
| Hyslop et al., 2016 [36] | HVJ-E | Heterologous PNT: approx. 39% Lower than non-manipulated controls (63%) and autologous PNT (60%) | Not tested | Embryos (modified technique): <5% (<2% in 79%; 2–5% in 21%) ESCs: <2% in 4/5 0–60% in 1/5 | Early PNT and modified manipulation medium. Higher quality blastocysts |
| Zhang et al., 2016 [35] | EF | 4-cell embryo: 71.4% (5/7) | Implantation: 60% (3/5) Births: 0% deaths by umbilical cord collapse | Fetal red cells: undetectable (undetermined sensitivity) | Reported a 2003 case |
| Wu et al., 2017 [37] | HVJ-E | 25.64% (13/64) similar to non-manipulated controls | Not tested | Embryos: 1 ± 1.45% ESCs: 0.26 ± 0.17% (P5-P20) | Employed pre-pronuclei without cytoskeleton disruption |
| Ref. | Species | Transfer Method | Blastocyst Develop | Births Rate | Carry-Over | Notes | |
|---|---|---|---|---|---|---|---|
| Wang et al., 2014 [31] | Mice (NZW and BDF1) | HVJ-E | PB1T | 82.4% (14/17) similar to MST | 42.8% (6/14) similar to MST | F1 tail: 0% (0/6) F2 fingers: 0% (0/16) lower to MST | Compared PB1T with MST and PB2T with PNT |
| PB2T | 40% (6/15) lower than PNT | 40% (6/15) similar to PNT | F1tail: 1.7 ± 2.8% F2 fingers: 2.9 ± 4.3% lower than PNT | ||||
| Ma et al., 2017 [39] | Humans | HVJ-E | PB1T | 32.5% (8/25) lower than control | Not tested | Not tested | Same group of refs: [19,30,33] |
| Zhang et al., 2017 [38] | Humans | HVJ-E | PB1T | 24.1% (7/29) similar to control | Not tested | Not tested | Data of fresh and cryogenized karyoplast transfers were added up |
| PB2T | 0% (0/17) lower than control | Not tested | |||||
| Wu et al., 2017 [40] | Humans | HVJ-E | PB1T | 25.3% (19/75) similar to control and PB2T | Not tested | Embryos: 0.26% (0.07–0.5%) ESCs: 0.15% (P5-P20) | Good results due to performing the transfer right after the fertilization (pre-pronucleus) Same group of ref. [37] |
| PB2T | 27.5% (14/51) similar to control and PB1T | Not tested | Embryos: 0.37% (0.06–0.7%) ESCs: 0.22% (P5-P20) | ||||
| MRT | Advantages | Disadvantages |
|---|---|---|
| GVT |
|
|
| MST |
|
|
| PNT |
|
|
| PB1T |
|
|
| PB2T |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sendra, L.; García-Mares, A.; Herrero, M.J.; Aliño, S.F. Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 551. https://doi.org/10.3390/ijms22020551
Sendra L, García-Mares A, Herrero MJ, Aliño SF. Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. International Journal of Molecular Sciences. 2021; 22(2):551. https://doi.org/10.3390/ijms22020551
Chicago/Turabian StyleSendra, Luis, Alfredo García-Mares, María José Herrero, and Salvador F. Aliño. 2021. "Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases" International Journal of Molecular Sciences 22, no. 2: 551. https://doi.org/10.3390/ijms22020551
APA StyleSendra, L., García-Mares, A., Herrero, M. J., & Aliño, S. F. (2021). Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. International Journal of Molecular Sciences, 22(2), 551. https://doi.org/10.3390/ijms22020551