
Redox Homeostasis and Regulation in Pluripotent Stem Cells: Uniqueness or Versatility?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Redox Homeostasis in Pluripotent Stem Cells
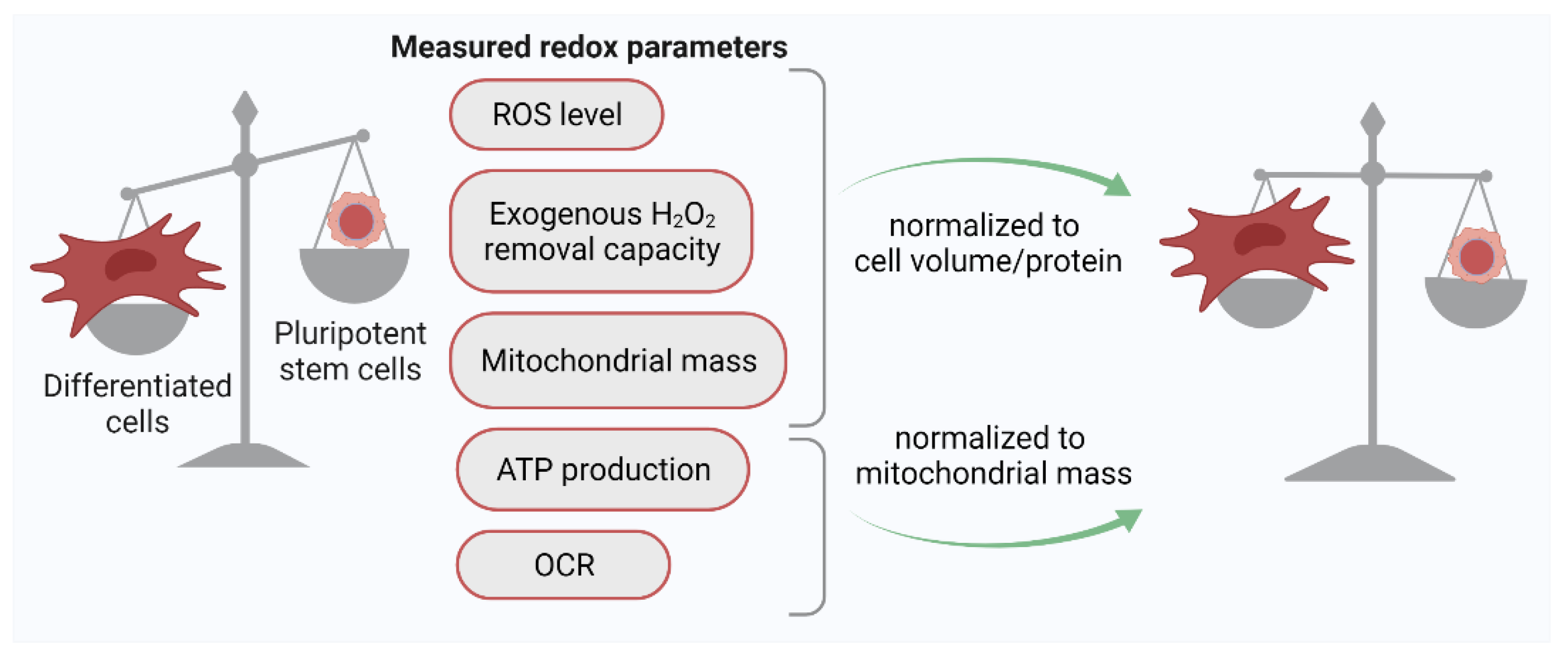
2.1. Quantification of the ROS Level in PSCs
2.2. ROS Production in PSCs
2.2.1. Mitochondrial Activity in PSCs
2.2.2. Non-Mitochondrial ROS Production in PSCs
2.3. ROS Elimination in PSCs
2.4. Oxidative Stress Response in PSCs
3. Redox Signaling in Pluripotent Stem Cells
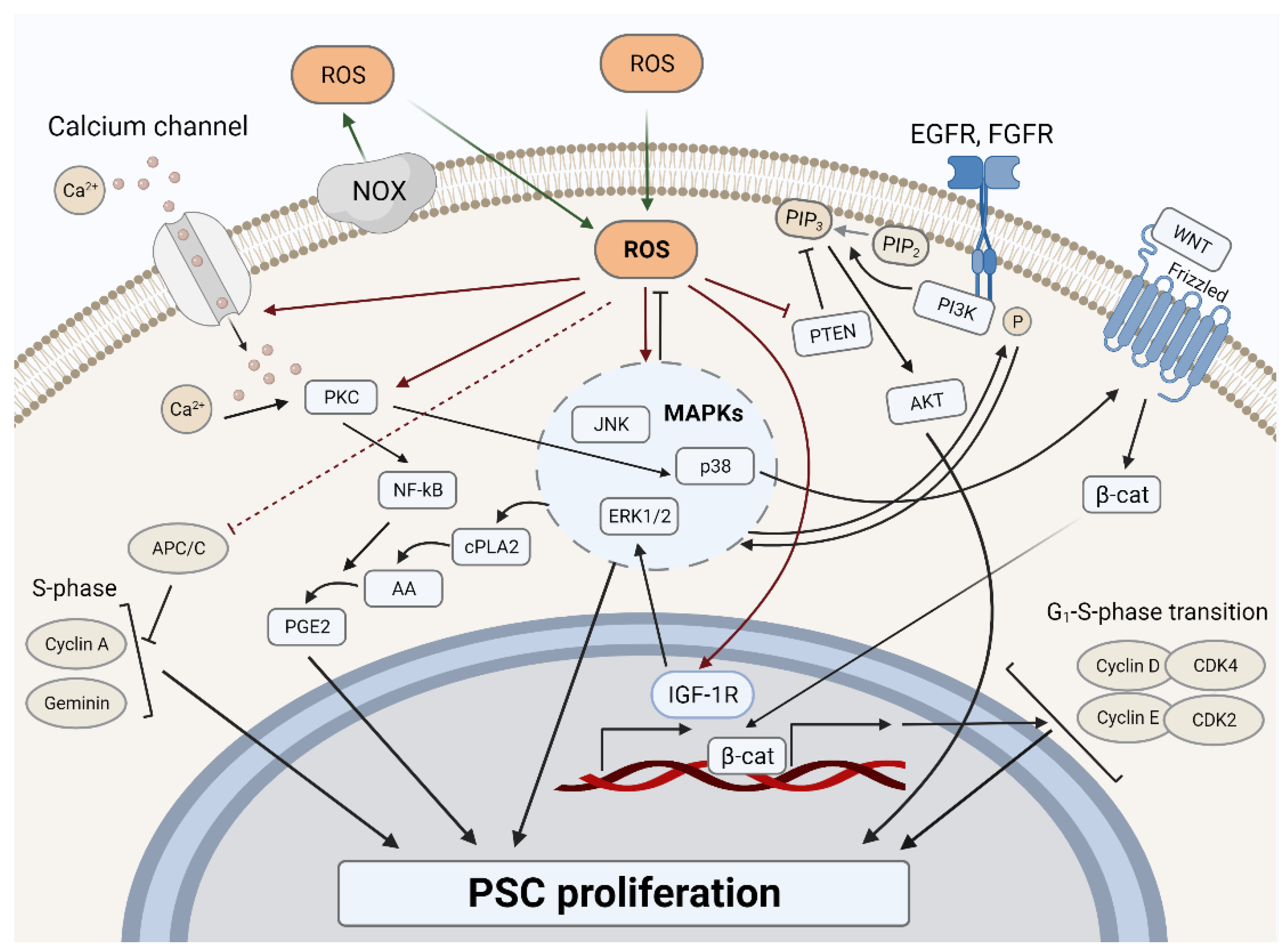
3.1. ROS and Proliferation in PSCs
3.1.1. ROS-Dependent Mitogenic Stimulation
3.1.2. ROS-Dependent Cell Cycle Regulation
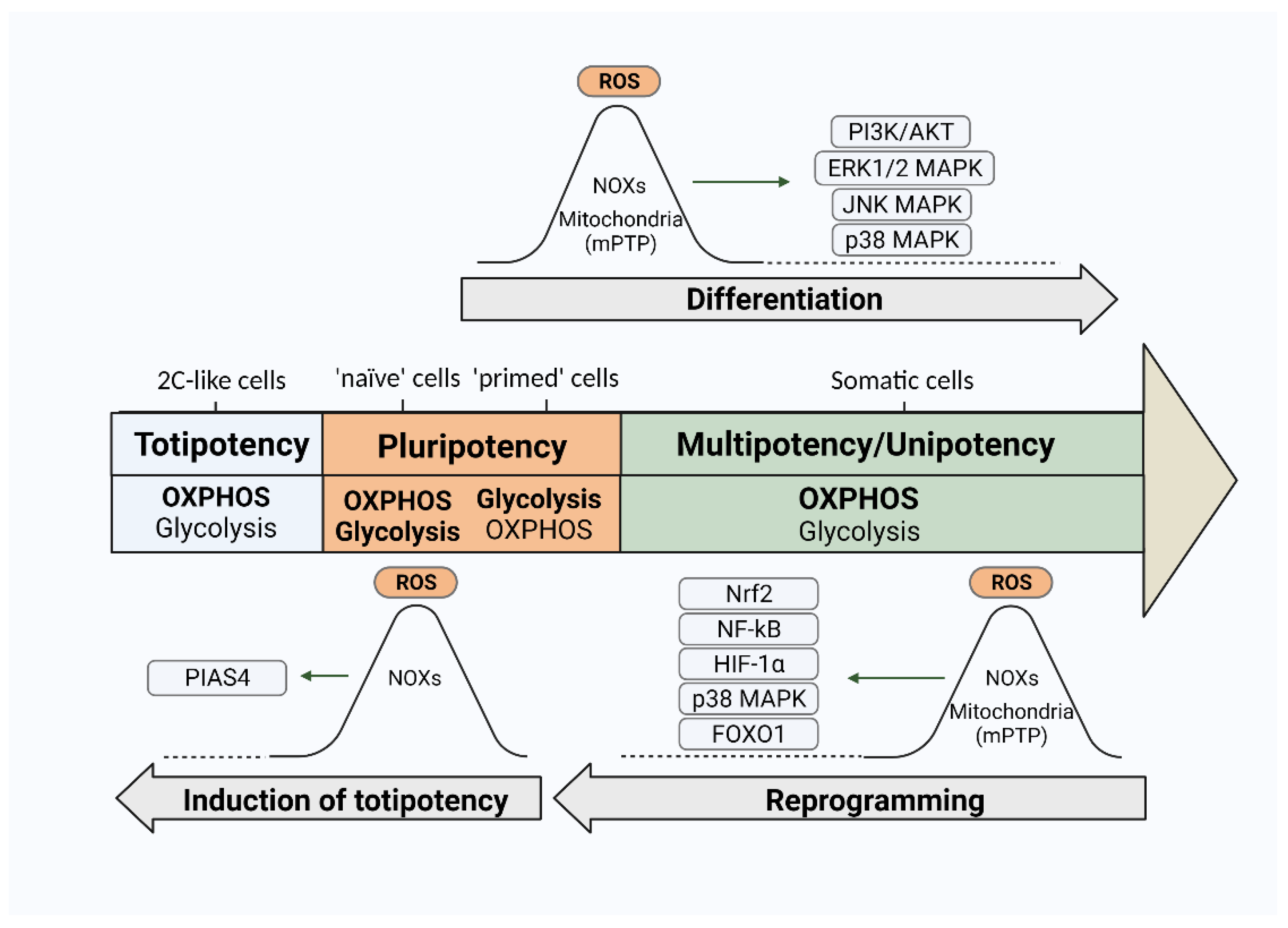
3.2. ROS and Differentiation of PSCs
3.3. ROS and Induction of Pluripotency
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Biological production, detection, and fate of hydrogen peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H. Biochemistry of Oxidative Stress. Angew. Chemie Int. Ed. English 1986, 25, 1058–1071. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Schmidt, H.H.H.W.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhubl, S.R. Why Have Antioxidants Failed in Clinical Trials? Am. J. Cardiol. 2008, 101, S14–S19. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilic, D.; Ogilvie, C. Concise Review: Human Embryonic Stem Cells—What Have We Done? What Are We Doing? Where Are We Going? Stem Cells 2017, 35, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boward, B.; Wu, T.; Dalton, S. Concise Review: Control of Cell Fate Through Cell Cycle and Pluripotency Networks. Stem Cells 2016, 34, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondoh, H.; Lleonart, M.E.; Nakashima, Y.; Yokode, M.; Tanaka, M.; Bernard, D.; Gil, J.; Beach, D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid. Redox Signal. 2007, 9, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A., IV; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.; Quek, L.E.; Titmarsh, D.; Krömer, J.O.; Kao, L.P.; Nielsen, L.; Wolvetang, E.; Cooper-White, J. Metabolic profiling and flux analysis of MEL-2 human embryonic stem cells during exponential growth at physiological and atmospheric oxygen concentrations. PLoS ONE 2014, 9, e112757. [Google Scholar] [CrossRef] [PubMed]
- Vlaski-Lafarge, M.; Loncaric, D.; Perez, L.; Labat, V.; Debeissat, C.; Brunet de la Grange, P.; Rossignol, R.; Ivanovic, Z.; Bœuf, H. Bioenergetic Changes Underline Plasticity of Murine Embryonic Stem Cells. Stem Cells 2019, 37, 463–475. [Google Scholar] [CrossRef]
- Saretzki, G. Stress Defense in Murine Embryonic Stem Cells Is Superior to That of Various Differentiated Murine Cells. Stem Cells 2004, 22, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Lyublinskaya, O.G.; Ivanova, J.S.; Pugovkina, N.A.; Kozhukharova, I.V.; Kovaleva, Z.V.; Shatrova, A.N.; Aksenov, N.D.; Zenin, V.V.; Kaulin, Y.A.; Gamaley, I.A.; et al. Redox environment in stem and differentiated cells: A quantitative approach. Redox Biol. 2017, 12, 758–769. [Google Scholar] [CrossRef]
- Ryu, J.M.; Lee, H.J.; Jung, Y.H.; Lee, K.H.; Kim, D.I.; Kim, J.Y.; Ko, S.H.; Choi, G.E.; Chai, I.I.; Song, E.J.; et al. Regulation of Stem Cell Fate by ROS-mediated Alteration of Metabolism. Int. J. Stem Cells 2015, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Kostyuk, A.I.; Panova, A.S.; Kokova, A.D.; Kotova, D.A.; Maltsev, D.I.; Podgorny, O.V.; Belousov, V.V.; Bilan, D.S. In vivo imaging with genetically encoded redox biosensors. Int. J. Mol. Sci. 2020, 21, 8164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, M.; Yuan, Z. Methods for the detection of reactive oxygen species. Anal. Methods 2018, 10, 4625–4638. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of Multiple Stress Defense Mechanisms During Differentiation of Human Embryonic Stem Cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2006, 348, 1472–1478. [Google Scholar] [CrossRef]
- Zhou, G.; Meng, S.; Li, Y.; Ghebre, Y.T.; Cooke, J.P. Optimal ROS Signaling Is Critical for Nuclear Reprogramming. Cell Rep. 2016, 15, 919–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, L.; Tilgner, K.; Saretzki, G.; Atkinson, S.P.; Stojkovic, M.; Moreno, R.; Przyborski, S.; Lako, M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010, 28, 661–673. [Google Scholar] [CrossRef]
- Forsyth, N.R.; Kay, A.; Hampson, K.; Downing, A.; Talbot, R.; McWhir, J. Transcriptome alterations due to physiological normoxic (2% O2) culture of human embryonic stem cells. Regen. Med. 2008, 3, 817–833. [Google Scholar] [CrossRef]
- Kigawa, J. Studies on the levels of pO2 and pCO2 in the uterine cavity and uterine tissue. Acta Obstet. Gynaecol. Jpn. 1981, 33, 1646–1654. [Google Scholar]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef]
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 5th ed.; W. H. Freeman & Co.: New York, NY, USA, 2008; 1119p. [Google Scholar]
- Varum, S.; Momčilović, O.; Castro, C.; Ben-Yehudah, A.; Ramalho-Santos, J.; Navara, C.S. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res. 2009, 3, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Arnold, P.K.; Rosen, B.P.; Chen, Y.; Carey, B.W.; Huangfu, D.; Carmona-Fontaine, C.; Thompson, C.B.; Finley, L.W.S. Glutamine independence is a selectable feature of pluripotent stemcells. Nat. Metab. 2019, 1, 676. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maraldi, T.; Angeloni, C.; Prata, C.; Hrelia, S. Nadph oxidases: Redox regulators of stem cell fate and function. Antioxidants 2021, 10, 973. [Google Scholar] [CrossRef]
- Forman, H.J.; Fukuto, J.M.; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 2004, 287, C246–C256. [Google Scholar] [CrossRef]
- Kawahara, Y.; Imanishi, T. A genome-wide survey of changes in protein evolutionary rates across four closely related species of Saccharomyces sensu stricto group. BMC Evol. Biol. 2007, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lalucque, H.; Silar, P. NADPH oxidase: An enzyme for multicellularity? Trends Microbiol. 2003, 11, 9–12. [Google Scholar] [CrossRef]
- Bedard, K.; Lardy, B.; Krause, K.H. NOX family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Compartmentalization of redox signaling through NaDPH oxidase-derived rOS. Antioxid. Redox Signal. 2009, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. Pharmacology & Therapeutics Regulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Wei, X.; Jiang, L.; Niu, C.; Zhang, J.; Chen, S.; Meng, D. Nox2 and Nox4 regulate self-renewal of murine induced-pluripotent stem cells. IUBMB Life 2016, 68, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Buggisch, M.; Ateghang, B.; Ruhe, C.; Strobel, C.; Lange, S.; Wartenberg, M.; Sauer, H. Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J. Cell Sci. 2007, 120, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Luo, Z.; Pepe, A.E.; Margariti, A.; Zeng, L.; Xu, Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am. J. Physiol. Cell Physiol. 2009, 296, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöcker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2013, 18, 1165. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, B.; Miura, T.; Brandenberger, R.; Mejido, J.; Luo, Y.; Yang, A.X.; Joshi, B.H.; Ginis, I.; Thies, R.S.; Amit, M.; et al. Gene expression in human embryonic stem cell lines: Unique molecular signature. Blood 2004, 103, 2956–2964. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Q.; Habegger, L.; Noisa, P.; Szekely, A.; Qiu, C.; Hutchison, S.; Raha, D.; Egholm, M.; Lin, H.; Weissman, S.; et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 5254–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, N.-Y.; Chan, Y.-S.; Feng, B.; Lu, X.; Orlov, Y.L.; Moreau, D.; Kumar, P.; Yang, L.; Jiang, J.; Lau, M.-S.; et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 2010, 468, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.J.; Laurent, L.C.; Kostka, D.; Ulitsky, I.; Williams, R.; Lu, C.; Park, I.H.; Rao, M.S.; Shamir, R.; Schwartz, P.H.; et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature 2008, 455, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Som, A. RNA-Seq analysis reveals pluripotency-associated genes and their interaction networks in human embryonic stem cells. Comput. Biol. Chem. 2020, 85, 107239. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef]
- Fan, J.; Robert, C.; Jang, Y.Y.; Liu, H.; Sharkis, S.; Baylin, S.B.; Rassool, F.V. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 713, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.L.; Chakraborty, S.; Rajan, S.S.; Wang, R.; Huang, F. Effects of oxidative stress on mouse embryonic stem cell proliferation, apoptosis, senescence, and self-renewal. Stem Cells Dev. 2010, 19, 1321–1331. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Pugovkina, N.A.; Zemelko, V.I.; Nikolsky, N.N.; Burova, E.B. Differences in defense mechanisms against oxidative stress in both human embryonic and endometrium-derived mesenchymal stem cells. Tsitologiya 2013, 55, 517–526. [Google Scholar]
- Jeong, A.Y.; Lee, M.Y.; Lee, S.H.; Park, J.H.; Han, H.J. PPARδ agonist-mediated ROS stimulates mouse embryonic stem cell proliferation through cooperation of p38 MAPK and Wnt/βcatenin. Cell Cycle 2009, 8, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Na, S.I.; Heo, J.S.; Kim, M.H.; Kim, Y.H.; Lee, M.Y.; Kim, S.H.; Lee, Y.J.; Han, H.J. Arachidonic acid release by H2O2 mediated proliferation of mouse embryonic stem cells: Involvement of Ca2+/PKC and mapks-induced EGFR transactivation. J. Cell. Biochem. 2009, 106, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Nugud, A.; Sandeep, D.; El-Serafi, A.T. Two faces of the coin: Minireview for dissecting the role of reactive oxygen species in stem cell potency and lineage commitment. J. Adv. Res. 2018, 14, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kučera, J.; Binó, L.; Štefková, K.; Jaroš, J.; Vašíček, O.; Večeřa, J.; Kubala, L.; Pacherník, J. Apocynin and diphenyleneiodonium induce oxidative stress and modulate PI3K/Akt and MAPK/Erk activity in mouse embryonic stem cells. Oxid. Med. Cell. Longev. 2016, 2016, 7409196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, P.; Ye, Z.; Jang, Y.-Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 2014, 20, 1881–1890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of Late G1/S Phase Transition and APCCdh1 by Reactive Oxygen Species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhans, W.C.; Heintz, N.H. The cell cycle is a redox cycle: Linking phase-speci fi c targets to cell fate. Free Radic. Biol. Med. 2009, 47, 1282–1293. [Google Scholar] [CrossRef]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [Green Version]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Kim, S.; Chung, H.; Pae, H. Reactive Oxygen Species in the Activation of MAP Kinases, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 528, ISBN 9780124058811. [Google Scholar]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarugi, P.; Buricchi, F. Protein tyrosine phosphorylation and reversible oxidation: Two cross-talking posttranslation modifications. Antioxid. Redox Signal. 2007, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Östman, A.; Frijhoff, J.; Sandin, Å.; Böhmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 2011, 150, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Downes, C.P. PTEN: The down side of PI 3-kinase signalling. Cell Signal. 2002, 14, 285–295. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.A.; Barbier, V.; Fotedar, A.; Fotedar, R. DNA damage triggers p21 WAF1-dependent emil down-regulation that maintains G2 arrest. Mol. Biol. Cell 2009, 20, 1891–1902. [Google Scholar] [CrossRef] [Green Version]
- Fojtík, P.; Beckerová, D.; Holomková, K.; Šenfluk, M.; Rotrekl, V. Both Hypoxia-Inducible Factor 1 and MAPK Signaling Pathway Attenuate PI3K/AKT via Suppression of Reactive Oxygen Species in Human Pluripotent Stem Cells. Front. Cell Dev. Biol. 2021, 8, 1784. [Google Scholar] [CrossRef]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Kernan, J.; Bonacci, T.; Emanuele, M.J. Who guards the guardian? Mechanisms that restrain APC/C during the cell cycle. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1924–1933. [Google Scholar] [CrossRef]
- Lyublinskaya, O.G.; Borisov, Y.G.; Pugovkina, N.A.; Smirnova, I.S.; Obidina, J.V.; Ivanova, J.S.; Zenin, V.V.; Shatrova, A.N.; Borodkina, A.V.; Aksenov, N.D.; et al. Reactive oxygen species are required for human mesenchymal stem cells to initiate proliferation after the quiescence exit. Oxid. Med. Cell. Longev. 2015, 2015, 502105. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent notch signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, J.D.R.; Freed, E.; Hsu, J.Y.; Kramer, E.R.; Peters, J.M.; Jackson, P.K. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 2001, 105, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Barta, T.; Dolezalova, D.; Holubcova, Z.; Hampl, A. Cell cycle regulation in human embryonic stem cells: Links to adaptation to cell culture. Exp. Biol. Med. 2013, 238, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Zhang, X.; Atkinson, S.; Lako, M. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene 2009, 28, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neganova, I.; Vilella, F.; Atkinson, S.P.; Lloret, M.; Passos, J.F.; Von Zglinicki, T.; O’Connor, J.E.; Burks, D.; Jones, R.; Armstrong, L.; et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells 2011, 29, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Ballabeni, A.; Park, I.H.; Zhao, R.; Wang, W.; Lerou, P.H.; Daley, G.Q.; Kirschner, M.W. Cell cycle adaptations of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19252–19257. [Google Scholar] [CrossRef] [Green Version]
- Rapkine, L. Sur les processus chimiques au cours de la division cellulaire—III.—Inhibition et rétablissement de la division cellulaire. J. Chim. Phys. 1937, 34, 416–427. [Google Scholar] [CrossRef]
- Kawamura, N. TRACE: Tennessee Research and Creative Exchange Studies of Protein-bound Sulfhydryl and Disulfide Groups in the Mitotic Apparatus of the Sea Urchin, Arbacia punctulata. Ph.D. Thesis, University of Tennessee, Knoxville, TN, USA, 1960. [Google Scholar]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca 2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, J.S.; Pugovkina, N.A.; Neganova, I.E.; Kozhukharova, I.V.; Nikolsky, N.N.; Lyublinskaya, O.G. Cell cycle-coupled changes in the level of reactive oxygen species support the proliferation of human pluripotent stem cells. Stem Cells 2021, in press. [Google Scholar] [CrossRef]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sánchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, H.; Rahimi, G.; Wartenberg, M. Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett. 2000, 476, 218–223. [Google Scholar] [CrossRef]
- Liang, J.; Wu, M.; Chen, C.; Mai, M.; Huang, J.; Zhu, P.; Agnetti, G. Roles of Reactive Oxygen Species in Cardiac Differentiation, Reprogramming, and Regenerative Therapies. Oxid. Med. Cell. Longev. 2020, 2020, 2102841. [Google Scholar] [CrossRef]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yan, Y.L.; Hao, J.; Wang, Y. Cellular redox state as a critical factor in initiating early embryonic-like program in embryonic stem cells. Cell Discov. 2019, 5, 1–4. [Google Scholar] [CrossRef]
- Urao, N.; Ushio-Fukai, M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic. Biol. Med. 2013, 54, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Wei, X.; Wang, X.; Jiang, L.; Niu, C.; Zhang, J.; Chen, S.; Meng, D. Nox2 contributes to the arterial endothelial specification of mouse induced pluripotent stem cells by upregulating Notch signaling. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Schmelter, M.; Ateghang, B.; Helmig, S.; Wartenberg, M.; Sauer, H.; Schmelter, M.; Ateghang, B.; Helmig, S.; Wartenberg, M.; Sauer, H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J. 2006, 20, 1182–1184. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid. Redox Signal. 2005, 7, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Serena, E.; Figallo, E.; Tandon, N.; Cannizzaro, C.; Gerecht, S.; Elvassore, N.; Vunjak-Novakovic, G. Electrical stimulation of human embryonic stem cells: Cardiac differentiation and the generation of reactive oxygen species. Exp. Cell Res. 2009, 315, 3611–3619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashkovskaia, N.; Gey, U.; Rödel, G. Mitochondrial ROS direct the differentiation of murine pluripotent P19 cells. Stem Cell Res. 2018, 30, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Crespo, F.L.; Sobrado, V.R.; Gomez, L.; Cervera, A.M.; McCreath, K.J. Mitochondrial Reactive Oxygen Species Mediate Cardiomyocyte Formation from Embryonic Stem Cells in High Glucose. Stem Cells 2010, 28, 1132–1142. [Google Scholar] [CrossRef]
- Cho, S.W.; Park, J.; Heo, H.J.; Park, S.; Song, S.; Kim, I.; Han, Y.; Yamashita, J.K.; Youm, J.B.; Han, J.; et al. Dual Modulation of the Mitochondrial Permeability Transition Pore and Redox Signaling Synergistically Promotes Cardiomyocyte Differentiation From Pluripotent Stem Cells. J. Am. Heart Assoc. 2014, 3, e000693. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Cong, X. The effect of reactive oxygen species on cardiomyocyte differentiation of pluripotent stem cells. Free Radic. Res. 2018, 52, 150–158. [Google Scholar] [CrossRef]
- Sart, S.; Song, L.; Li, Y. Controlling redox status for stem cell survival, expansion, and differentiation. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, T.V.A.; Smyrnias, I.; Shah, A.M.; Brewer, A.C. NADPH oxidase 4 regulates cardiomyocyte differentiation via redox activation of c-Jun protein and the cis-Regulation of GATA-4 gene transcription. J. Biol. Chem. 2013, 288, 15745–15759. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, X.; Jiang, K.; Liu, J.; Liu, Z. Dural effects of oxidative stress on cardiomyogenesis via Gata4 transcription and protein ubiquitination. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.U.; Park, Y.H.; Kim, J.M.; Sun, H.N.; Song, I.S.; Huang, S.M.; Lee, S.H.; Chae, J.-I.; Hong, S.; Sik Choi, S.; et al. Dominant role of peroxiredoxin/JNK axis in stemness regulation during neurogenesis from embryonic stem cells. Stem Cells 2014, 32, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.R.; Ku, S.Y.; Cho, M.S.; Kim, Y.Y.Y.J.; Kim, Y.Y.Y.J.; Oh, S.K.; Kim, S.H.; Moon, S.Y.; Choi, Y.M. Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp. Mol. Med. 2010, 42, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Liang, X.G.; Hu, Y.; Zhu, D.Y.; Lou, Y.J. Involvement of p38MAPK and reactive oxygen species in icariin-induced cardiomyocyte differentiation of murine embryonic stem cells in vitro. Stem Cells Dev. 2008, 17, 751–760. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Polo, J.M. Phases of reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Prigione, A.; Rohwer, N.; Hoffmann, S.; Mlody, B.; Drews, K.; Bukowiecki, R.; Blümlein, K.; Wanker, E.E.; Ralser, M.; Cramer, T.; et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 2014, 32, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Lee, S.A.; Prasain, N.; Bae, D.; Kang, H.; Ha, T.; Kim, J.S.; Hong, K.S.; Mantel, C.; Moon, S.H.; et al. Metabolome Profiling of Partial and Fully Reprogrammed Induced Pluripotent Stem Cells. Stem Cells Dev. 2017, 26, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 1994. [Google Scholar]
- Kida, Y.S.; Kawamura, T.; Wei, Z.; Sogo, T.; Jacinto, S.; Shigeno, A.; Kushige, H.; Yoshihara, E.; Liddle, C.; Ecker, J.R.; et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell 2015, 16, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; Ponsoda, X.; Izpisua Belmonte, J.C.; Torres, J. Mitochondrial dynamics and metabolism in induced pluripotency. Exp. Gerontol. 2020, 133, 110870. [Google Scholar] [CrossRef] [PubMed]
- Hansson, J.; Rafiee, M.R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly Coordinated Proteome Dynamics during Reprogramming of Somatic Cells to Pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Z.; Xiang, G.; Zheng, L.; Tang, H.; Duan, L.; Lin, X.; Zhao, Q.; Chen, K.; Wu, Y.; Xing, G.; et al. Short-Term Mitochondrial Permeability Transition Pore Opening Modulates Histone Lysine Methylation at the Early Phase of Somatic Cell Reprogramming. Cell Metab. 2018, 28, 935–945.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 Orchestrates the Metabolic Shift during Induced Pluripotent Stem Cell Reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Sharma, V.; Qi, S.; Guarch, M.E.; Zhao, P.; Luo, Z.; Fan, W.; Wang, Y.; Mbabaali, F.; Neculai, D.; et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Ruohola-Baker, H. Metabolic remodeling during the loss and acquisition of pluripotency. Development 2017, 144, 541. [Google Scholar] [CrossRef] [Green Version]
- Neganova, I.; Chichagova, V.; Armstrong, L.; Lako, M. A critical role for p38MAPK signalling pathway during reprogramming of human fibroblasts to iPSCs. Sci. Rep. 2017, 7, 41693. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yalcin, S.; Lee, D.F.; Yeh, T.Y.J.; Lee, S.M.; Su, J.; Mungamuri, S.K.; Rimmelé, P.; Kennedy, M.; Sellers, R.; et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Matsuda, M.; Kawamura, T.; Sogo, T.; Shigeno, A.; Nishida, E.; Ebisuya, M. Foxd1 is a mediator and indicator of the cell reprogramming process. Nat. Commun. 2014, 5, 3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, J.S.; Lyublinskaya, O.G. Redox Homeostasis and Regulation in Pluripotent Stem Cells: Uniqueness or Versatility? Int. J. Mol. Sci. 2021, 22, 10946. https://doi.org/10.3390/ijms222010946
Ivanova JS, Lyublinskaya OG. Redox Homeostasis and Regulation in Pluripotent Stem Cells: Uniqueness or Versatility? International Journal of Molecular Sciences. 2021; 22(20):10946. https://doi.org/10.3390/ijms222010946
Chicago/Turabian StyleIvanova, Julia S., and Olga G. Lyublinskaya. 2021. "Redox Homeostasis and Regulation in Pluripotent Stem Cells: Uniqueness or Versatility?" International Journal of Molecular Sciences 22, no. 20: 10946. https://doi.org/10.3390/ijms222010946
APA StyleIvanova, J. S., & Lyublinskaya, O. G. (2021). Redox Homeostasis and Regulation in Pluripotent Stem Cells: Uniqueness or Versatility? International Journal of Molecular Sciences, 22(20), 10946. https://doi.org/10.3390/ijms222010946