
Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention


Abstract
:1. Introduction
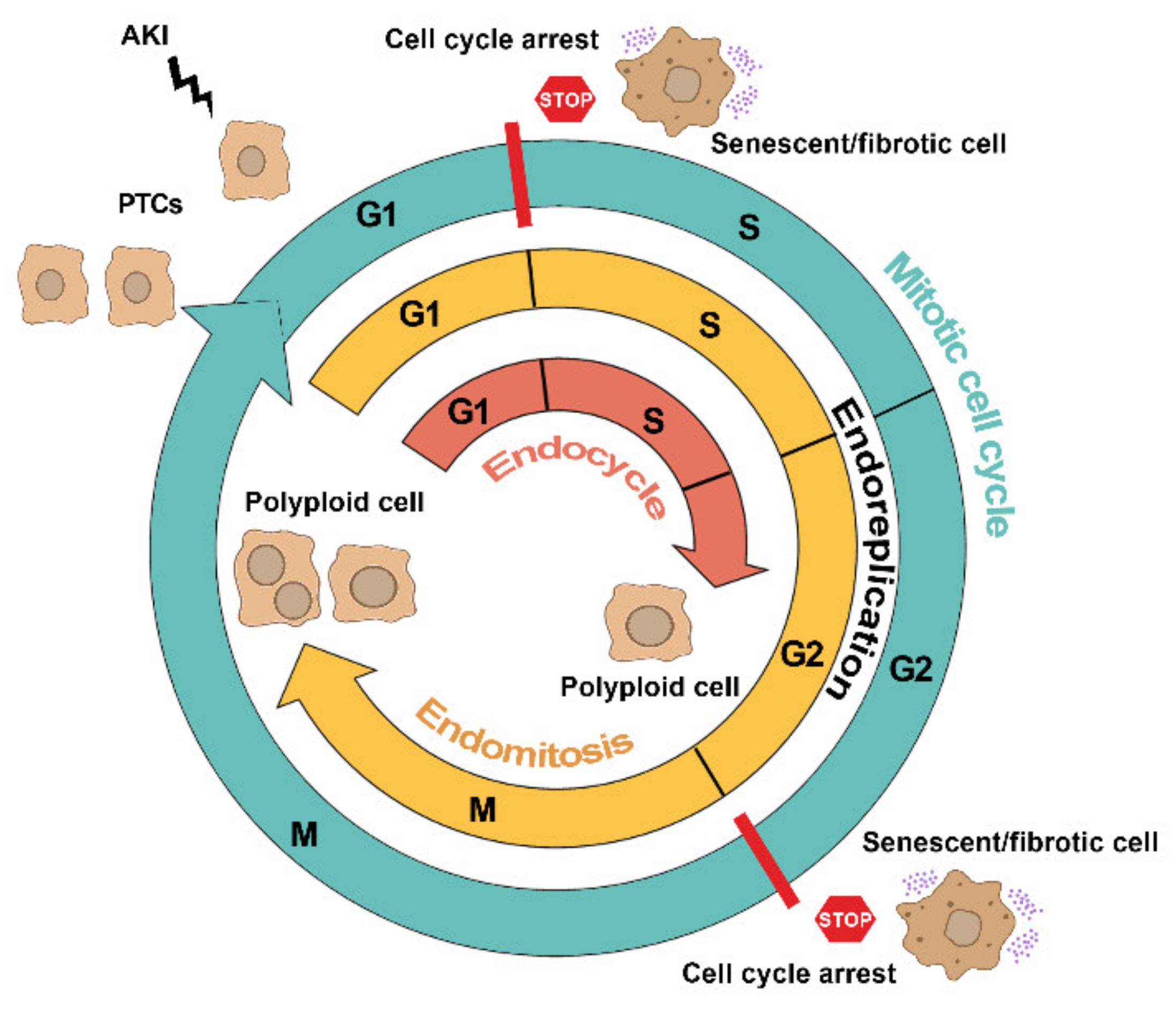
2. The Many Faces of Cell Cycle in Tubular Cells after AKI
2.1. Mitotic Cell Cycle
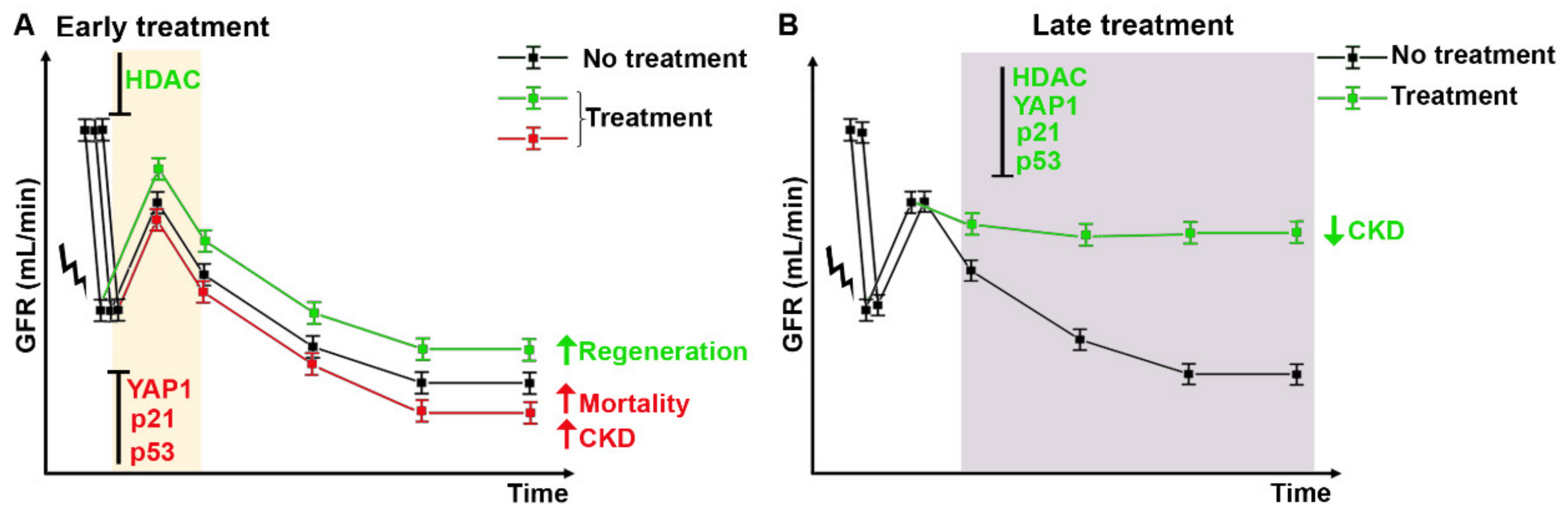
Targeting Mitotic Cell Cycle as Potential Innovative Strategy for CKD Prevention
- HDAC inhibitors
2.2. Cell Cycle Arrest
Targeting Cell Cycle Arrest as Potential Innovative Strategy for CKD Prevention
- p21Waf1/Cip1
- p53
- HDAC inhibitors
2.3. Alternative Cell Cycles
Targeting Alternative Cell Cycle as Potential Innovative Strategy for CKD Prevention
- YAP1

{kind=link}
{kind=link}
{kind=link}
| Cell Cycle Phase | Target | Therapeutic Strategy | Timing of Treatment | Effect | Reference | |
|---|---|---|---|---|---|---|
| G1/S | HDAC inhibitor (4-PBA) | 1 day after IRI | Renal function recovery and tubular regeneration | [23] | ||
| Mitotic cell cycle | G1/S | HDAC | HDAC inhibitor (TSA) | 1 day after IRI | Renal function recovery and tubular regeneration | [23] |
| G1/S | HDAC inhibitor (m4PTB) | 1 day after injury | Renal function improvement and fibrosis decrease | [43] | ||
| G1 | p21 | p21(−/−) mouse, constitutive KO | - | Increase of tubular cell death and mortality | [58] | |
| G1 | p21(−/−) mouse, constitutive KO | - | No fibrosis development | [62] | ||
| G2 | p21(−/−) mouse, constitutive KO | - | Fibrosis exacerbation | [63] | ||
| G1 | Cdk2 inhibitor (Purvalanol) | 1 day after injury | Nephrotoxicity reduction | [60] | ||
| G1 | Cdk4/6 inhibitor (PD 0332991) | 1 h before IRI | Renal inflammation attenuation and kidney damage improvement | [42] | ||
| Cell cycle arrest | G1 | Smad7(−/−) mouse, constitutive KO | - | Tubular regeneration impairment | [64] | |
| G2/M | p53 inhibitor (Pifithrin-a) | On the day of IRI | Fibrosis increase | [68] | ||
| G2/M | p53 | MDM2 antagonist (Nutlin-3a) | 1 day before injury | Renal inflammation and tubular injury decrease | [71] | |
| G2/M | p53 inhibitor (Pifithrin-a) | 3 days after injury | Fibrosis decrease | [47] | ||
| G2/M | JNK inhibitor (SP600125) | 7 days after injury | Fibrosis decrease | [47] | ||
| G1/S | HDAC | HDAC inhibitor (UPHD186) | 3 days after injury | Fibrosis decrease | [75] | |
| G1/S | HDAC inhibitor (m4PTB) | 4 days after injury | Fibrosis decrease | [76] | ||
| G1/S | HDAC inhibitor (UPHD186) | 4 days after injury | Fibrosis decrease | [77] | ||
| G1/S | HDAC6 inhibitor (ACY-1215) | On the day of injury | Fibrosis decrease | [78] | ||
| Alternative cell cycle | G1/S | YAP1 | YAP1(−/−) mouse, renal conditional KO | - | Delay of renal function recovery | [96] |
| G1/S | YAP1 inhibitor (Verteporfin) | On the day of IRI | Delay of renal function recovery | [96] | ||
| - | YAP1 silencing (Ad-shYAP) | 7 days after IRI | Renal function recovery and fibrosis decrease | [93] | ||
| - | KLF4 silencing (Ad-shKLF4) | 7 days after IRI | Renal function recovery and fibrosis decrease | [93] | ||
| - | KLF4 overexpression (Ad-KLF4) | 7 days after IRI | Fibrosis increase | [93] | ||
| G2/M | YAP1 inhibitor (Verteporfin) | 3 days after IRI | Renal inflammation and fibrosis decrease | [94] |
- The Notch pathway
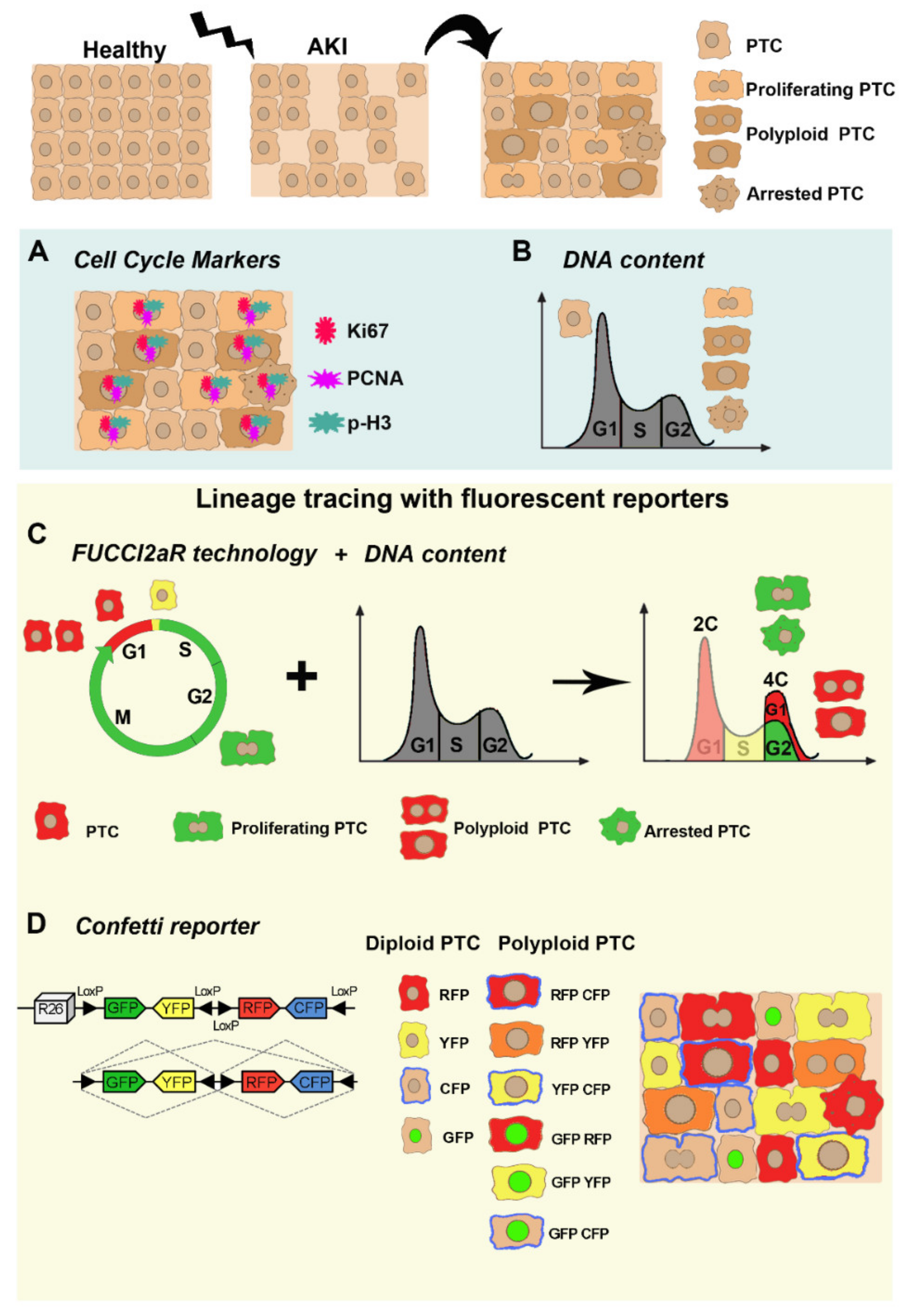
3. Mitotic Cell Cycle, Cell Cycle Arrest, or Alternative Cell Cycle: How Can We Get to the Bottom?
3.1. Cell Cycle Markers
3.2. DNA Content Analysis
3.3. Lineage Tracing with Fluorescent Reporters
4. Biological Significance of PTC Cell Cycle Behavior
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKI | acute kidney injury |
| CKD | chronic kidney disease |
| PTC | proximal tubular cells |
| ESKD | end-stage kidney disease |
| G0, G1, G2 phases | gap 0, gap 1, gap 2 phases |
| S phase | synthesis Phase |
| M phase | mitosis phase |
| DNA | deoxyribonucleic acid |
| PCNA | proliferating cell nuclear antigen |
| BrdU | bromodeoxyuridine |
| RPC | renal progenitor cells |
| HDAC | histone deacetylase |
| m4PTB | methyl-4-(phenylthio) butanoate |
| TSA | trichostatin |
| 4-PBA | 4-phenylbutyrate |
| ATM | ataxia telangiectasia mutated |
| ATR | ataxia telangiectasia and Rad3-related protein |
| CHK2 | checkpoint kinase 2 |
| TIMP2 | tissue inhibitor of metalloproteinase 2 |
| IGFBP7 | insulin-like growth factor-binding protein 7 |
| TGFβ | transforming growth factor beta |
| CTGF | connective tissue growth factor |
| JNK | c-jun NH2- terminal kinase |
| MAPK | mitogen-activated protein kinase |
| SASP | senescence-associated secretory phenotype |
| snRNA-seq | single nucleus cell RNA-seq |
| p-H3+ | phospho-histone H3+ |
| CDK | cyclin dependent kinase |
| Smad | small mother against decapentaplegic |
| UUO | unilateral ureteral obstruction |
| PTBA | phenylthiobutanoic acids |
| RPTEC | Renal proximal tubular epithelial cells |
| YAP1 | yes-associated protein 1 |
| CDH1/FZR1 | fizzy and cell division cycle 20 related 1 |
| APC/C | anaphase-promoting complex/cyclosome |
| LATS | large tumor suppressor |
| E2F | E2F Transcription Factor |
| NICD | notch intracellular domain |
| EdU | 3H-thymidine, 5-ethynyl-20-deoxyuridine |
| FUCCI2aR | fluorescent ubiquitination-based cell cycle indicator |
| Cdt1 | chromatin licensing and DNA replication factor1 |
| GFP | green fluorescent protein |
| YFP | yellow fluorescent protein |
| RFP | red fluorescent protein |
| CFP | cyan fluorescent protein |
| KO | knock-out |
| IRI | ischemia reperfusion injury |
References
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Kottgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Perico, N.; Remuzzi, G. Acute kidney injury: More awareness needed, globally. Lancet 2015, 386, 1425–1427. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Hsu, R.K.; Hsu, C.Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Nakhoul, N.; Batuman, V. Role of proximal tubules in the pathogenesis of kidney disease. Contrib. Nephrol. 2011, 169, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Schuh, C.D.; Polesel, M.; Platonova, E.; Haenni, D.; Gassama, A.; Tokonami, N.; Ghazi, S.; Bugarski, M.; Devuyst, O.; Ziegler, U.; et al. Combined Structural and Functional Imaging of the Kidney Reveals Major Axial Differences in Proximal Tubule Endocytosis. J. Am. Soc. Nephrol. 2018, 29, 2696–2712. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Endou, H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am. J. Physiol. 1988, 255, F977–F983. [Google Scholar] [CrossRef]
- Lieberthal, W.; Nigam, S.K. Acute renal failure. I. Relative importance of proximal vs. distal tubular injury. Am. J. Physiol. 1998, 275, F623–F631. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chang, J.; Yao, B.; Niu, A.; Kelly, E.; Breeggemann, M.C.; Abboud Werner, S.L.; Harris, R.C.; Zhang, M.Z. Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int. 2015, 88, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, S.C.; Huynh, L.; Marlier, A.; Lee, Y.; Moeckel, G.W.; Cantley, L.G. GM-CSF Promotes Macrophage Alternative Activation after Renal Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1334–1345. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Bonventre, J.V. Acute Kidney Injury and Progression of Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Gembillo, G.; Cernaro, V.; Siligato, R.; Curreri, F.; Catalano, A.; Santoro, D. Protective Role of Vitamin D in Renal Tubulopathies. Metabolites 2020, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Prim. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2015, 30, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Liu, J.; Pang, P.; Krautzberger, A.M.; Reginensi, A.; Akiyama, H.; Schedl, A.; Humphreys, B.D.; McMahon, A.P. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell. Rep. 2015, 12, 1325–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell. Stem. Cell. 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang-Panesso, M.; Kadyrov, F.F.; Lalli, M.; Wu, H.; Ikeda, S.; Kefaloyianni, E.; Abdelmageed, M.M.; Herrlich, A.; Kobayashi, A.; Humphreys, B.D. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J. Clin. Investig. 2019, 129, 5501–5517. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.; Bangen, J.M.; Hammerich, L.; Liedtke, C.; Floege, J.; Smeets, B.; Moeller, M.J. Origin of regenerating tubular cells after acute kidney injury. Proc. Natl. Acad. Sci. USA 2014, 111, 1533–1538. [Google Scholar] [CrossRef] [Green Version]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem. Cells 2012, 30, 1714–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagrinati, C.; Netti, G.S.; Mazzinghi, B.; Lazzeri, E.; Liotta, F.; Frosali, F.; Ronconi, E.; Meini, C.; Gacci, M.; Squecco, R.; et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J. Am. Soc. Nephrol. 2006, 17, 2443–2456. [Google Scholar] [CrossRef] [Green Version]
- Rinkevich, Y.; Montoro, D.T.; Contreras-Trujillo, H.; Harari-Steinberg, O.; Newman, A.M.; Tsai, J.M.; Lim, X.; Van-Amerongen, R.; Bowman, A.; Januszyk, M.; et al. In vivo clonal analysis reveals lineage-restricted progenitor characteristics in mammalian kidney development, maintenance, and regeneration. Cell. Rep. 2014, 7, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, P.; Rinkevich, Y.; Dekel, B. The use of lineage tracing to study kidney injury and regeneration. Nat. Rev. Nephrol. 2015, 11, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Smeets, B.; Boor, P.; Dijkman, H.; Sharma, S.V.; Jirak, P.; Mooren, F.; Berger, K.; Bornemann, J.; Gelman, I.H.; Floege, J.; et al. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J. Pathol. 2013, 229, 645–659. [Google Scholar] [CrossRef]
- Appel, D.; Kershaw, D.B.; Smeets, B.; Yuan, G.; Fuss, A.; Frye, B.; Elger, M.; Kriz, W.; Floege, J.; Moeller, M.J. Recruitment of podocytes from glomerular parietal epithelial cells. J. Am. Soc. Nephrol. 2009, 20, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Rookmaaker, M.B.; Kujala, P.; Ng, A.; Leushacke, M.; Snippert, H.; van de Wetering, M.; Tan, S.; Van Es, J.H.; Huch, M.; et al. Lgr5(+ve) stem/progenitor cells contribute to nephron formation during kidney development. Cell. Rep. 2012, 2, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell. Stem. Cell. 2008, 3, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Lasagni, L.; Angelotti, M.L.; Ronconi, E.; Lombardi, D.; Nardi, S.; Peired, A.; Becherucci, F.; Mazzinghi, B.; Sisti, A.; Romoli, S.; et al. Podocyte Regeneration Driven by Renal Progenitors Determines Glomerular Disease Remission and Can Be Pharmacologically Enhanced. Stem. Cell. Reports 2015, 5, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, D.; Bostrom, A.K.; Nilsson, K.; Hansson, J.; Sjolund, J.; Moller, C.; Jirstrom, K.; Nilsson, E.; Landberg, G.; Axelson, H.; et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am. J. Pathol. 2011, 178, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiRocco, D.P.; Bisi, J.; Roberts, P.; Strum, J.; Wong, K.K.; Sharpless, N.; Humphreys, B.D. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F379–F388. [Google Scholar] [CrossRef] [Green Version]
- Cianciolo Cosentino, C.; Skrypnyk, N.I.; Brilli, L.L.; Chiba, T.; Novitskaya, T.; Woods, C.; West, J.; Korotchenko, V.N.; McDermott, L.; Day, B.W.; et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 2013, 24, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomasova, D.; Anders, H.J. Cell cycle control in the kidney. Nephrol. Dial. Transplant. 2015, 30, 1622–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Kellum, J.A.; Chawla, L.S. Cell-cycle arrest and acute kidney injury: The light and the dark sides. Nephrol. Dial. Transplant. 2016, 31, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Kashani, K.; Al-Khafaji, A.; Ardiles, T.; Artigas, A.; Bagshaw, S.M.; Bell, M.; Bihorac, A.; Birkhahn, R.; Cely, C.M.; Chawla, L.S.; et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013, 17, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 2009, 76, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Preisig, P.A.; Franch, H.A. Renal epithelial cell hyperplasia and hypertrophy. Semin. Nephrol. 1995, 15, 327–340. [Google Scholar]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- de Borst, M.H.; Prakash, J.; Sandovici, M.; Klok, P.A.; Hamming, I.; Kok, R.J.; Navis, G.; van Goor, H. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J. Pharmacol. Exp. Ther. 2009, 331, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell. Commun. Signal 2018, 12, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Matjusaitis, M.; Chin, G.; Sarnoski, E.A.; Stolzing, A. Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 2016, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, L.M.S.; Liu, J.; Koppitch, K.; Cippa, P.E.; McMahon, A.P. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2026684118. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Andrade, L.; Vieira, J.M., Jr.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakura, T.; Fujigaki, Y.; Fujikura, T.; Ohashi, N.; Kato, A.; Yasuda, H. Acquired resistance to rechallenge injury after acute kidney injury in rats is associated with cell cycle arrest in proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2016, 310, F872–F884. [Google Scholar] [CrossRef] [Green Version]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Price, P.M.; Tamayo, E.; Safirstein, R.L. The lack of a functional p21(WAF1/CIP1) gene ameliorates progression to chronic renal failure. Proc. Natl. Acad. Sci. USA 1999, 96, 10830–10835. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Tarcsafalvi, A.; Li, S.; Hodeify, R.; Seng, N.S.; Portilla, D.; Price, P.M. Increased expression of p21WAF1/CIP1 in kidney proximal tubules mediates fibrosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F122–F130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyano, T.; Namba, M.; Kobayashi, T.; Nakakuni, K.; Nakano, D.; Fukushima, M.; Nishiyama, A.; Matsuyama, M. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 2019, 9, 12059. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Tang, Y.; Huang, X.R.; Feng, M.; Xu, A.P.; Lan, H.Y. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the G1 cell cycle arrest. Clin. Sci. 2017, 131, 1955–1969. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Dagher, P.C.; Sandoval, R.M.; Campos, S.B.; Ashush, H.; Fridman, E.; Brafman, A.; Faerman, A.; Atkinson, S.J.; Thompson, J.D.; et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Lai, K.N.; Lan, H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010, 21, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-beta1/p53 signaling in renal fibrogenesis. Cell. Signal 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dagher, P.C.; Mai, E.M.; Hato, T.; Lee, S.Y.; Anderson, M.D.; Karozos, S.C.; Mang, H.E.; Knipe, N.L.; Plotkin, Z.; Sutton, T.A. The p53 inhibitor pifithrin-alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F284–F291. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, J.H.; Thomasova, D.; Mulay, S.R.; Anders, H.J. Nrf2 signalling promotes ex vivo tubular epithelial cell survival and regeneration via murine double minute (MDM)-2. Nephrol. Dial. Transplant. 2013, 28, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Anders, H.J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012, 81, 1199–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Kulkarni, O.P.; Migliorini, A.; Bruns, H.; Grobmayr, R.; Lazzeri, E.; Lasagni, L.; Liapis, H.; et al. Podocyte loss involves MDM2-driven mitotic catastrophe. J. Pathol. 2013, 230, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [Green Version]
- Skrypnyk, N.I.; Sanker, S.; Skvarca, L.B.; Novitskaya, T.; Woods, C.; Chiba, T.; Patel, K.; Goldberg, N.D.; McDermott, L.; Vinson, P.N.; et al. Delayed treatment with PTBA analogs reduces postinjury renal fibrosis after kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F705–F716. [Google Scholar] [CrossRef] [Green Version]
- Novitskaya, T.; McDermott, L.; Zhang, K.X.; Chiba, T.; Paueksakon, P.; Hukriede, N.A.; de Caestecker, M.P. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am. J. Physiol. Renal. Physiol. 2014, 306, F496–F504. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Li, S.; Frank, A.; Chen, X.; Emlet, D.; Hukriede, N.A.; Kellum, J.A. Time-dependent effects of histone deacetylase inhibition in sepsis-associated acute kidney injury. Intensive Care Med. Exp. 2020, 8, 9. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Hou, X.; Li, J.; Li, T.; Qiu, A.; Liu, N.; Zhuang, S. Histone deacetylase 6 inhibition mitigates renal fibrosis by suppressing TGF-beta and EGFR signaling pathways in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2020, 319, F1003–F1014. [Google Scholar] [CrossRef]
- Donne, R.; Saroul-Ainama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef]
- Ovrebo, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Manolopoulou, M.; Matlock, B.K.; Nlandu-Khodo, S.; Simmons, A.J.; Lau, K.S.; Phillips-Mignemi, M.; Ivanova, A.; Alford, C.E.; Flaherty, D.K.; Gewin, L.S. Novel kidney dissociation protocol and image-based flow cytometry facilitate improved analysis of injured proximal tubules. Am. J. Physiol. Ren. Physiol. 2019, 316, F847–F855. [Google Scholar] [CrossRef]
- Payne, E.H.; Ramalingam, D.; Fox, D.T.; Klotman, M.E. Polyploidy and Mitotic Cell Death Are Two Distinct HIV-1 Vpr-Driven Outcomes in Renal Tubule Epithelial Cells. J. Virol. 2018, 92, e01718-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Otto, E.A.; Cluckey, A.; Airik, R.; Hurd, T.W.; Chaki, M.; Diaz, K.; Lach, F.P.; Bennett, G.R.; Gee, H.Y.; et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012, 44, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Chipchase, M.D.; O’Neill, M.; Melton, D.W. Characterization of premature liver polyploidy in DNA repair (Ercc1)-deficient mice. Hepatology 2003, 38, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Apelt, K.; White, S.M.; Kim, H.S.; Yeo, J.E.; Kragten, A.; Wondergem, A.P.; Rooimans, M.A.; Gonzalez-Prieto, R.; Wiegant, W.W.; Lunke, S.; et al. ERCC1 mutations impede DNA damage repair and cause liver and kidney dysfunction in patients. J. Exp. Med. 2021, 218, e20200622. [Google Scholar] [CrossRef]
- Kim, W.; Cho, Y.S.; Wang, X.; Park, O.; Ma, X.; Kim, H.; Gan, W.; Jho, E.H.; Cha, B.; Jeung, Y.J.; et al. Hippo signaling is intrinsically regulated during cell cycle progression by APC/C(Cdh1). Proc. Natl. Acad. Sci. USA 2019, 116, 9423–9432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with purpose. Genes. Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.; Allen, S.R.; Sawyer, J.K.; Fox, D.T. Fizzy-Related dictates A cell cycle switch during organ repair and tissue growth responses in the Drosophila hindgut. Elife 2018, 7, e38327. [Google Scholar] [CrossRef] [PubMed]
- Kai, T.; Tsukamoto, Y.; Hijiya, N.; Tokunaga, A.; Nakada, C.; Uchida, T.; Daa, T.; Iha, H.; Takahashi, M.; Nomura, T.; et al. Kidney-specific knockout of Sav1 in the mouse promotes hyperproliferation of renal tubular epithelium through suppression of the Hippo pathway. J. Pathol. 2016, 239, 97–108. [Google Scholar] [CrossRef]
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37, e00565-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, E.; Kim, W.Y.; Hur, J.; Kim, H.; Nam, S.A.; Choi, A.; Kim, Y.M.; Park, S.H.; Chung, C.; Kim, J.; et al. The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Sci. Rep. 2016, 6, 31931. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, L.; Zhang, Y.; Li, W.; Li, J.; Wang, Y.; Meng, C.; Qin, J.; Zheng, Z.H.; Lan, H.Y.; et al. Tubule-Specific Mst1/2 Deficiency Induces CKD via YAP and Non-YAP Mechanisms. J. Am. Soc. Nephrol. 2020, 31, 946–961. [Google Scholar] [CrossRef]
- Xu, D.; Chen, P.P.; Zheng, P.Q.; Yin, F.; Cheng, Q.; Zhou, Z.L.; Xie, H.Y.; Li, J.Y.; Ni, J.Y.; Wang, Y.Z.; et al. KLF4 initiates sustained YAP activation to promote renal fibrosis in mice after ischemia-reperfusion kidney injury. Acta Pharmacol. Sin. 2021, 42, 436–450. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, C.; Shao, G.; Li, J.; Xu, K.; Zhao, Z.; Zhang, Z.; Liu, J.; Wu, H. Hippo-YAP/MCP-1 mediated tubular maladaptive repair promote inflammation in renal failed recovery after ischemic AKI. Cell. Death Dis. 2021, 12, 754. [Google Scholar] [CrossRef]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; You, H.; Li, Y.; Xu, Y.; He, Q.; Harris, R.C. EGF Receptor-Dependent YAP Activation Is Important for Renal Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 2372–2385. [Google Scholar] [CrossRef]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef] [Green Version]
- Von Stetina, J.R.; Frawley, L.E.; Unhavaithaya, Y.; Orr-Weaver, T.L. Variant cell cycles regulated by Notch signaling control cell size and ensure a functional blood-brain barrier. Development 2018, 145, dev157115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.M.; Althauser, C.; Ruohola-Baker, H. Notch-Delta signaling induces a transition from mitotic cell cycle to endocycle in Drosophila follicle cells. Development 2001, 128, 4737–4746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Yang, S.A.; Gong, S.; Chang, C.H.; Portilla, J.M.; Chatterjee, D.; Irianto, J.; Bao, H.; Huang, Y.C.; Deng, W.M. Polyploid mitosis and depolyploidization promote chromosomal instability and tumor progression in a Notch-induced tumor model. Dev. Cell. 2021, 56, 1976–1988.e4. [Google Scholar] [CrossRef] [PubMed]
- Peired, A.J.; Antonelli, G.; Angelotti, M.L.; Allinovi, M.; Guzzi, F.; Sisti, A.; Semeraro, R.; Conte, C.; Mazzinghi, B.; Nardi, S.; et al. Acute kidney injury promotes development of papillary renal cell adenoma and carcinoma from renal progenitor cells. Sci. Transl. Med. 2020, 12, eaaw6003. [Google Scholar] [CrossRef] [PubMed]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H.; et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, T.; Beck, A.R.; Rougier, N.; McNeil, T.; Lucian, L.; Werb, Z.; Amati, B. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J. 2003, 22, 4794–4803. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Zhou, Q.; Veeraragoo, P.; Yu, H.; Xiao, Z. Notch2/Hes-1 pathway plays an important role in renal ischemia and reperfusion injury-associated inflammation and apoptosis and the gamma-secretase inhibitor DAPT has a nephroprotective effect. Ren. Fail. 2011, 33, 207–216. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Conway, E.M.; Harris, R.C. Survivin mediates renal proximal tubule recovery from AKI. J. Am. Soc. Nephrol. 2013, 24, 2023–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Huang, S.; Reidy, K.; Han, S.H.; Chinga, F.; Susztak, K. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell. Rep. 2016, 14, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Richardson, G.D. Simultaneous Assessment of Cardiomyocyte DNA Synthesis and Ploidy: A Method to Assist Quantification of Cardiomyocyte Regeneration and Turnover. J. Vis. Exp. 2016, 53979. [Google Scholar] [CrossRef] [Green Version]
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 13409–13414. [Google Scholar] [CrossRef] [Green Version]
- Losick, V.P.; Jun, A.S.; Spradling, A.C. Wound-Induced Polyploidization: Regulation by Hippo and JNK Signaling and Conservation in Mammals. PLoS ONE 2016, 11, e0151251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, D.; Becherucci, F.; Romagnani, P. How much can the tubule regenerate and who does it? An open question. Nephrol. Dial. Transplant. 2016, 31, 1243–1250. [Google Scholar] [CrossRef] [Green Version]
- Mort, R.L.; Ford, M.J.; Sakaue-Sawano, A.; Lindstrom, N.O.; Casadio, A.; Douglas, A.T.; Keighren, M.A.; Hohenstein, P.; Miyawaki, A.; Jackson, I.J. Fucci2a: A bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell. Cycle. 2014, 13, 2681–2696. [Google Scholar] [CrossRef] [Green Version]
- Zielke, N.; Edgar, B.A. FUCCI sensors: Powerful new tools for analysis of cell proliferation. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative polyploid cells give rise to tumors via ploidy reduction. Nat. Commun. 2021, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wakefield, L.; Tarlow, B.D.; Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell. Stem Cell. 2020, 26, 34–47.e33. [Google Scholar] [CrossRef]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell. 2018, 47, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahouma, M.; Arisha, M.J.; Elmously, A.; El-Sayed Ahmed, M.M.; Spadaccio, C.; Mehta, K.; Baudo, M.; Kamel, M.; Mansor, E.; Ruan, Y.; et al. Cardiac tumors prevalence and mortality: A systematic review and meta-analysis. Int. J. Surg. 2020, 76, 178–189. [Google Scholar] [CrossRef]
- Gan, P.; Patterson, M.; Sucov, H.M. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu. Rev. Physiol. 2020, 82, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Wang, J.; Jackman, C.P.; Cox, A.H.; Trembley, M.A.; Balowski, J.J.; Cox, B.D.; De Simone, A.; Dickson, A.L.; Di Talia, S.; et al. Tension Creates an Endoreplication Wavefront that Leads Regeneration of Epicardial Tissue. Dev. Cell. 2017, 42, 600–615.e604. [Google Scholar] [CrossRef] [Green Version]
- Rios, A.C.; Fu, N.Y.; Jamieson, P.R.; Pal, B.; Whitehead, L.; Nicholas, K.R.; Lindeman, G.J.; Visvader, J.E. Essential role for a novel population of binucleated mammary epithelial cells in lactation. Nat. Commun. 2016, 7, 11400. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, P.K.; Zelnick, L.R.; Chinchilli, V.M.; Moledina, D.G.; Coca, S.G.; Parikh, C.R.; Garg, A.X.; Hsu, C.Y.; Go, A.S.; Liu, K.D.; et al. Association Between Early Recovery of Kidney Function After Acute Kidney Injury and Long-term Clinical Outcomes. JAMA Netw. Open. 2020, 3, e202682. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellum, J.A.; Sileanu, F.E.; Bihorac, A.; Hoste, E.A.; Chawla, L.S. Recovery after Acute Kidney Injury. Am. J. Respir. Crit. Care Med. 2017, 195, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Charlton, J.R.; Springsteen, C.H.; Carmody, J.B. Nephron number and its determinants in early life: A primer. Pediatr. Nephrol. 2014, 29, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Palant, C.E.; Mahajan, V.; Chawla, L.S. Sequelae of AKI. Best. Pract. Res. Clin. Anaesthesiol. 2017, 31, 415–425. [Google Scholar] [CrossRef]
- Silver, S.A.; Harel, Z.; McArthur, E.; Nash, D.M.; Acedillo, R.; Kitchlu, A.; Garg, A.X.; Chertow, G.M.; Bell, C.M.; Wald, R. Causes of Death after a Hospitalization with AKI. J. Am. Soc. Nephrol. 2018, 29, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; Haak, A.J.; Tschumperlin, D.J.; LeBrasseur, N.K. Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr. Rheumatol. Rep. 2018, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Xue, L.; Yang, F.; Dai, S.; Han, Z.; Liu, K.; Liu, B.; Yuan, Q.; Cui, Z.; Zhang, Y.; et al. Postinfarction Hearts Are Protected by Premature Senescent Cardiomyocytes Via GATA 4-Dependent CCN 1 Secretion. J. Am. Heart Assoc. 2018, 7, e009111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaud, G.; Brooks, C.R.; Kishi, S.; Taguchi, K.; Nishimura, K.; Magassa, S.; Scott, A.; Hsiao, L.L.; Ichimura, T.; Terzi, F.; et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Djudjaj, S.; Martin, I.V.; Buhl, E.M.; Nothofer, N.J.; Leng, L.; Piecychna, M.; Floege, J.; Bernhagen, J.; Bucala, R.; Boor, P. Macrophage Migration Inhibitory Factor Limits Renal Inflammation and Fibrosis by Counteracting Tubular Cell Cycle Arrest. J. Am. Soc. Nephrol. 2017, 28, 3590–3604. [Google Scholar] [CrossRef]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Chiara, L.; Conte, C.; Antonelli, G.; Lazzeri, E. Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention. Int. J. Mol. Sci. 2021, 22, 11093. https://doi.org/10.3390/ijms222011093
De Chiara L, Conte C, Antonelli G, Lazzeri E. Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention. International Journal of Molecular Sciences. 2021; 22(20):11093. https://doi.org/10.3390/ijms222011093
Chicago/Turabian StyleDe Chiara, Letizia, Carolina Conte, Giulia Antonelli, and Elena Lazzeri. 2021. "Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention" International Journal of Molecular Sciences 22, no. 20: 11093. https://doi.org/10.3390/ijms222011093
APA StyleDe Chiara, L., Conte, C., Antonelli, G., & Lazzeri, E. (2021). Tubular Cell Cycle Response upon AKI: Revising Old and New Paradigms to Identify Novel Targets for CKD Prevention. International Journal of Molecular Sciences, 22(20), 11093. https://doi.org/10.3390/ijms222011093