
Unraveling Protein Interactions between the Temperate Virus Bam35 and Its Bacillus Host Using an Integrative Yeast Two Hybrid–High Throughput Sequencing Approach

 , , ,
, , ,  , and
, and
Abstract
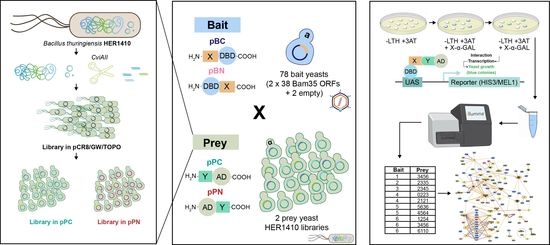
:
1. Introduction
2. Results
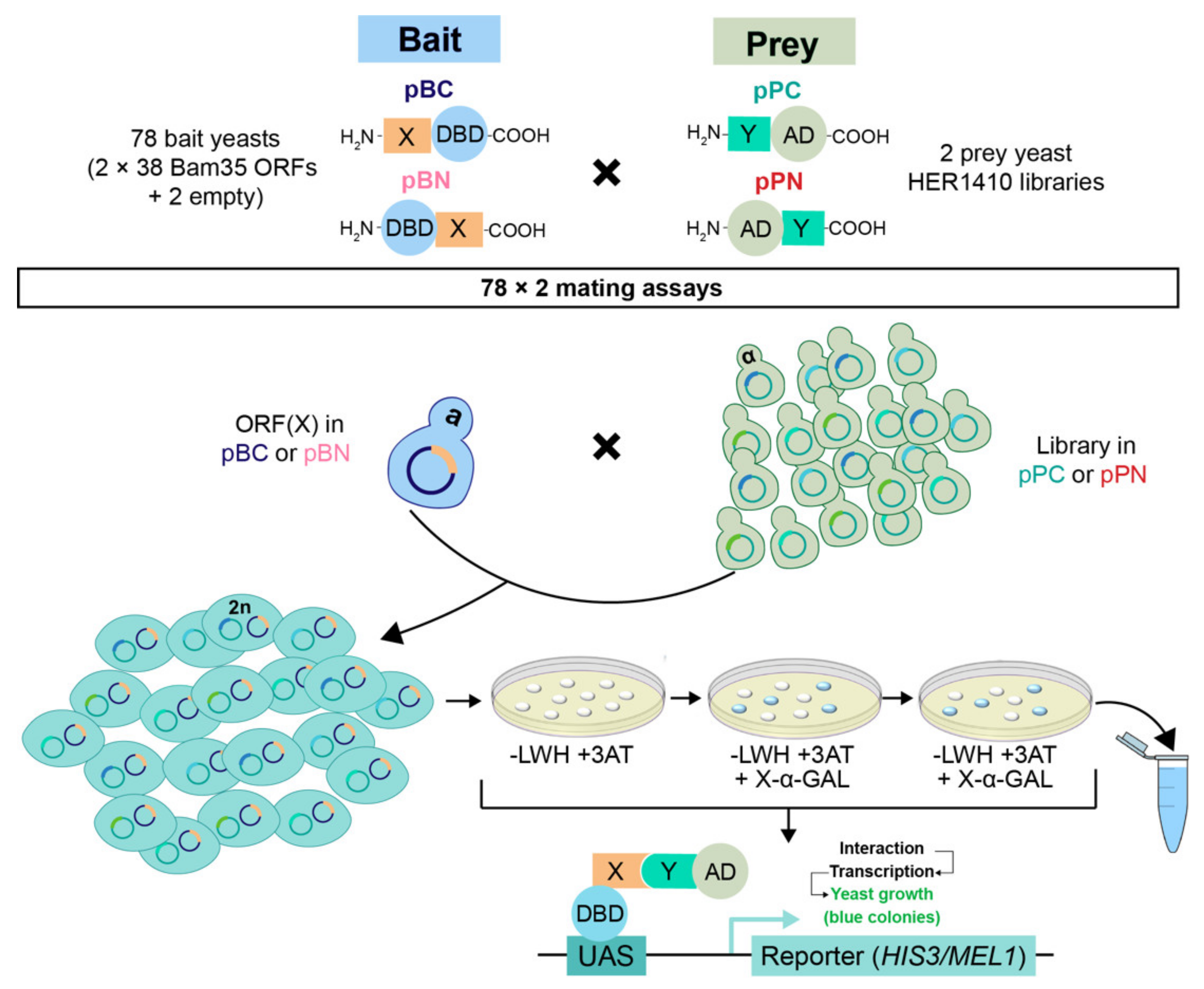
2.1. Integrating the Yeast Two-Hybrid System with high-throughput Sequencing for High-Confidence Interaction Datasets
2.2. Challenging Y2H Single Hits from the Fragment Genomic Library Using ORF Pairwise Y2H Assays
2.3. Bam35–B. thuringiensis Y2H-HTS Predicted the Interactome: Clear the Forest to Predict PPIs
3. Discussion
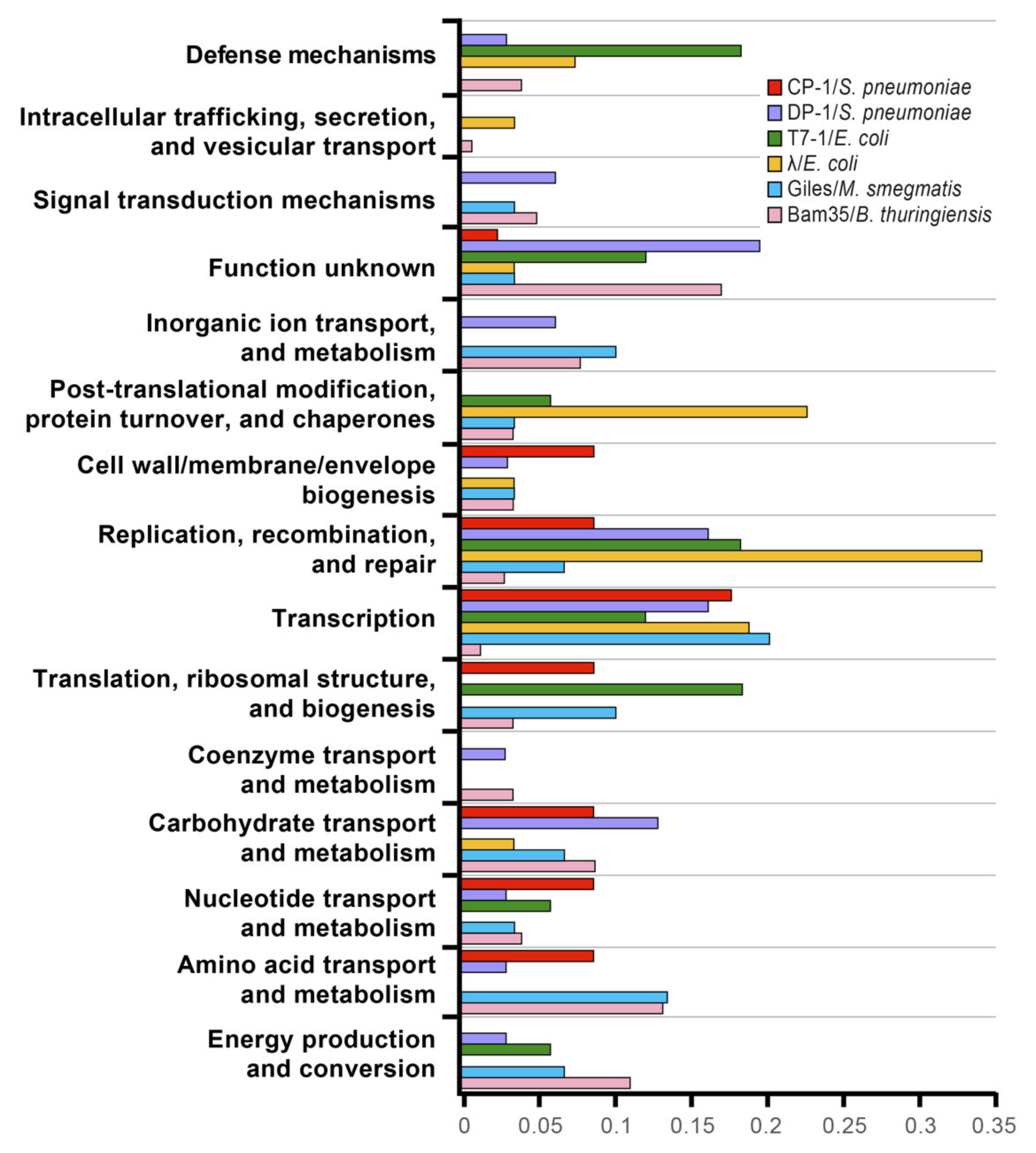
3.1. A Proficient Y2H-HTS Method to Detect Multiple Phage–Host PPIs
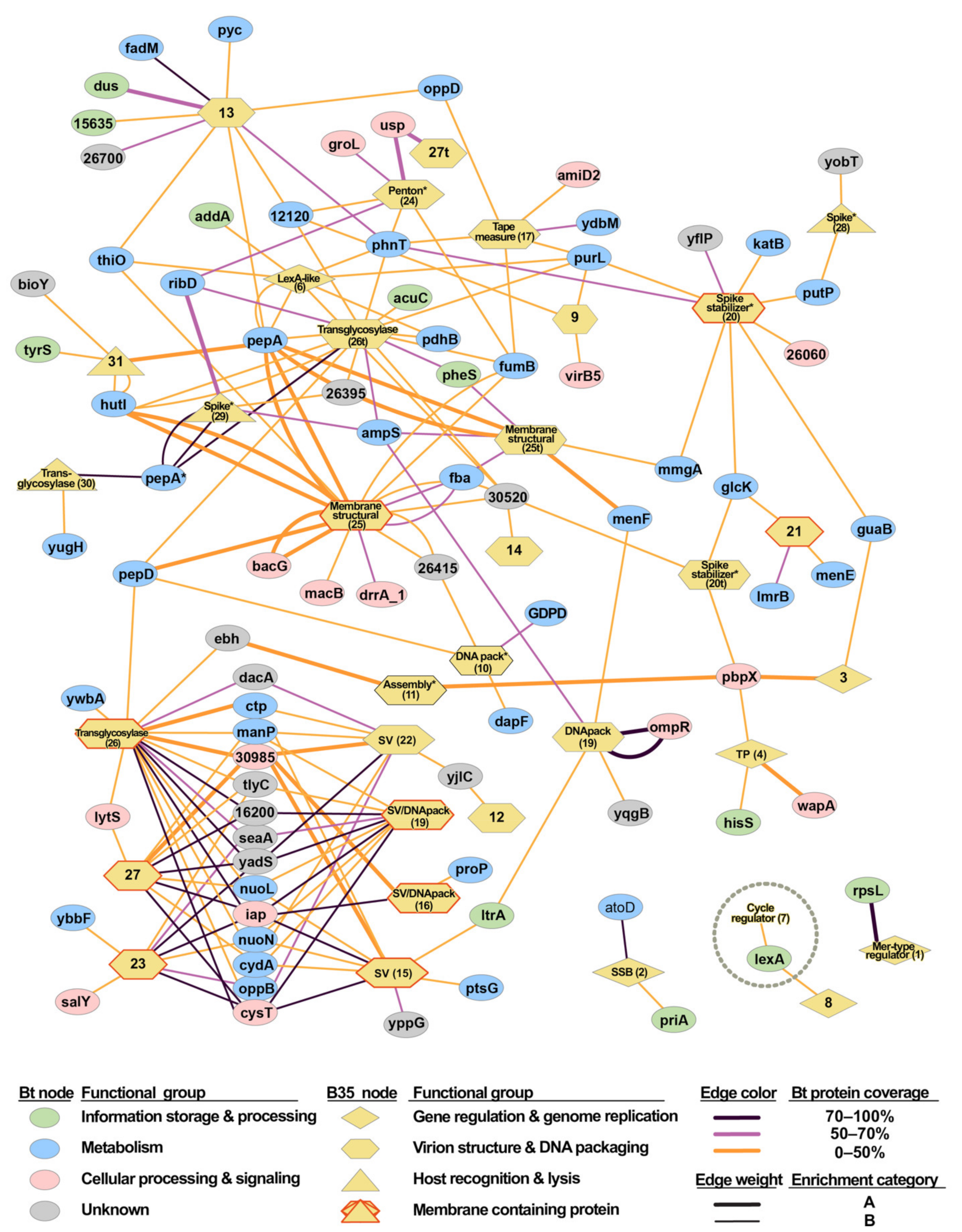
3.2. The Bam35–Bt Y2H Interactome Revealed the Clustering of Special Vertex Proteins and a Wide Modulation of Host Cell Metabolism
4. Materials and Methods
4.1. Nucleotides and DNAs
4.2. Bacterial and Yeast Strains
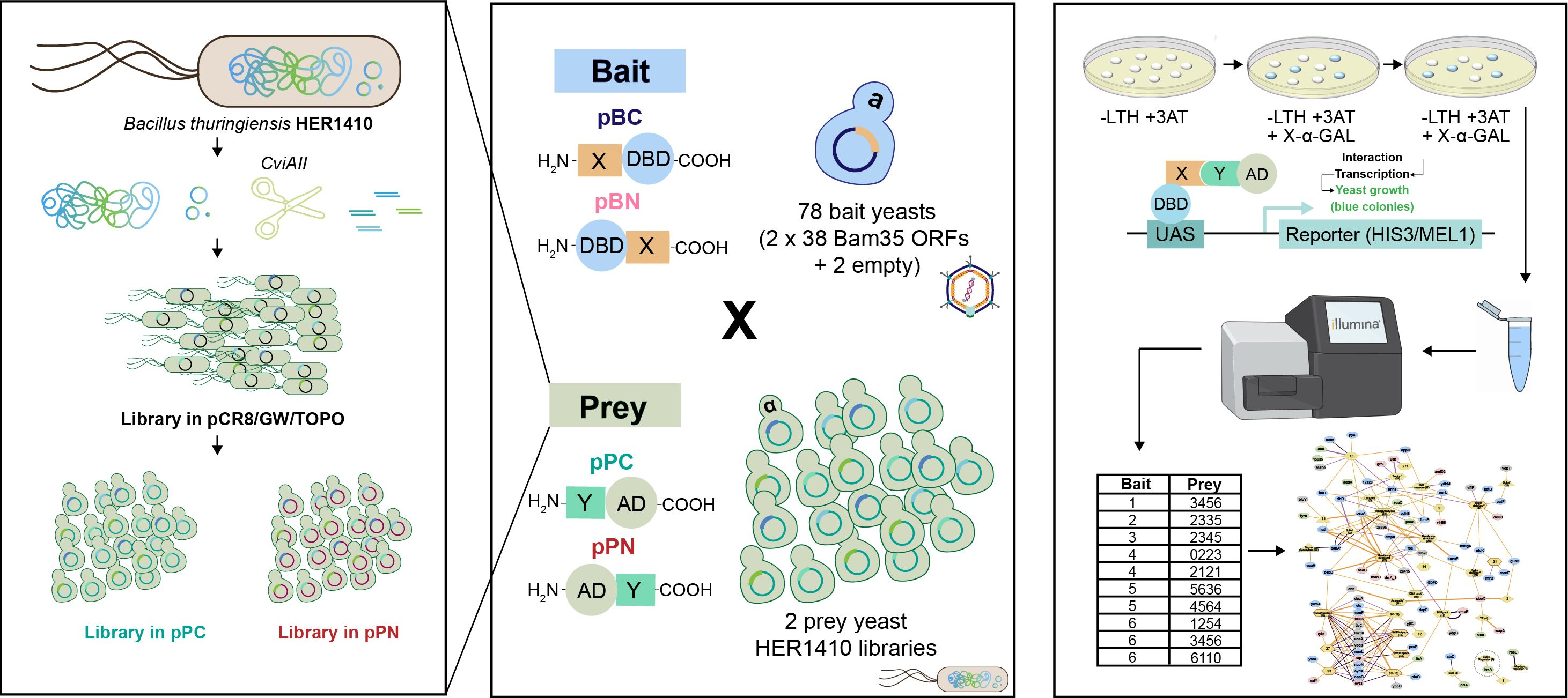
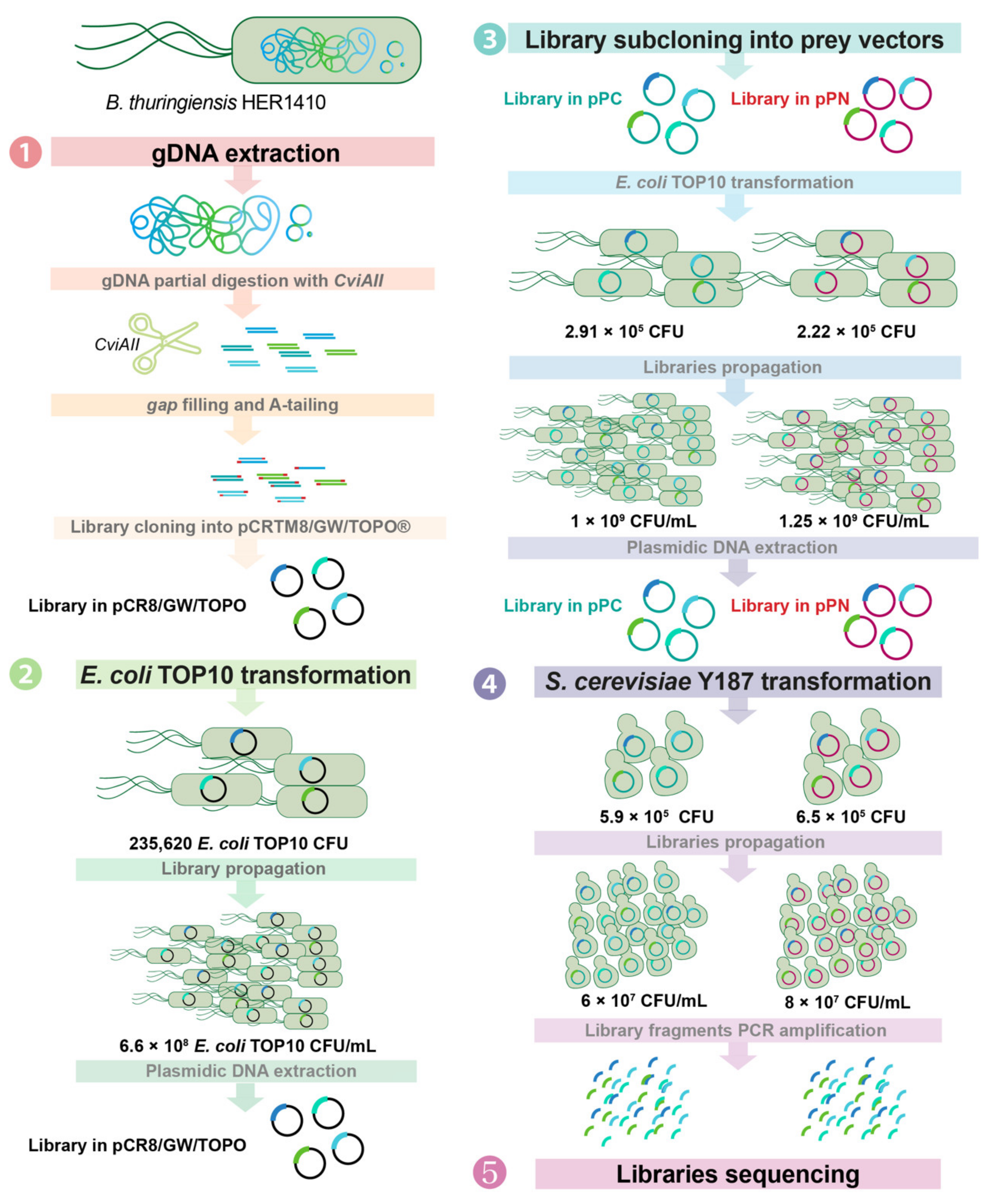
4.3. Genomic Library Construction
4.3.1. Generation of the Genomic Library and Cloning into the Donor Vector
4.3.2. Amplification of the Genomic Library in pCRTM8/GW/TOPO®
4.3.3. Subcloning of the Library in the Y2H Expression Vectors
4.3.4. Transformation of Saccharomyces cerevisiae Y187 with the Y2H Genomic Libraries
4.4. Yeast Two-Hybrid Screening
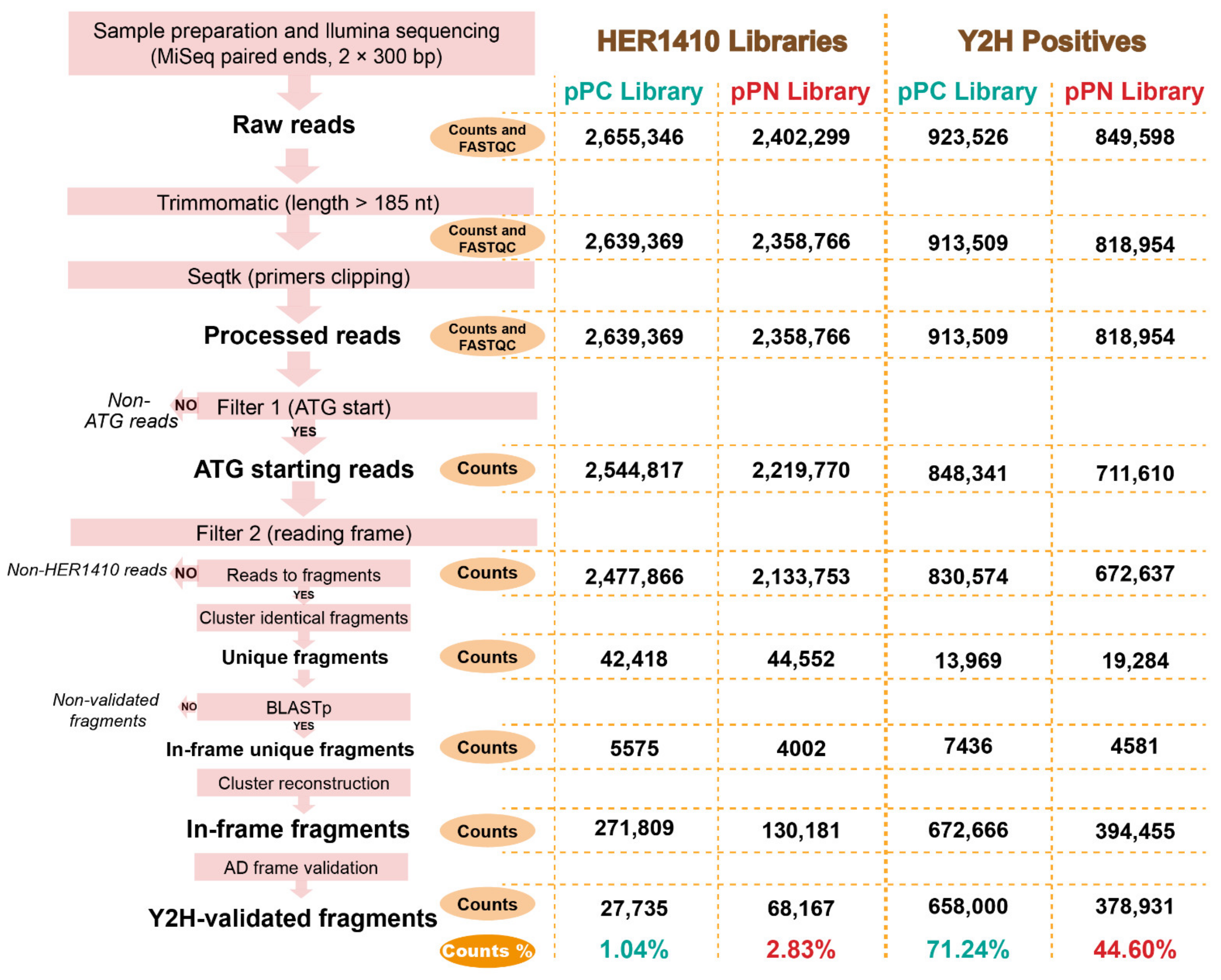
4.5. Genomic Library and Y2H Positives Analyses Using High-Throughput Sequencing
4.5.1. pPC and pPN Libraries Sequencing
4.5.2. Y2H Positives Preparation and Sequencing
4.5.3. Trimming, Quality Check, and Mapping of Illumina Reads
4.5.4. Evaluation of Genomic Library Quality
4.5.5. Evaluation of Raw Y2H Interactions
4.6. Full-Length Protein Pairwise Y2H Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oksanen, H.M.; Bamford, D.H. Family Tectiviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, 2012th ed.; Andrew, M.Q., King, M.J.A., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier, Academic Press: San Diego, CA, USA, 2012; Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 12 October 2021).
- Caruso, S.M.; deCarvalho, T.N.; Erill, I.; Gill, J.J.; Gillis, A. Taxonomic Proposal 2020.042B.R Create Two New Genera (Deltatectivirus and Epsilontectivirus) Including Three New Species (Kalamavirales: Tectiviridae). 2021. Available online: https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/prokaryote-official/12150 (accessed on 12 October 2021).
- Yutin, N.; Bäckström, D.; Ettema, T.J.G.; Krupovic, M.; Koonin, E.V. Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol. J. 2018, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.M.; Decarvalho, T.N.; Huynh, A.; Morcos, G.; Kuo, N.; Parsa, S.; Erill, I. A Novel Genus of Actinobacterial Tectiviridae. Viruses 2019, 11, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of Bacterial and Archaeal Viruses: Dynamics within the Prokaryotic Virosphere. Microbiol. Mol. Biol. Rev. 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupovic, M.; Koonin, E.V. Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 2014, 13, 105–115. [Google Scholar] [CrossRef]
- Jalasvuori, M.; Koskinen, K. Extending the hosts of Tectiviridae into four additional genera of Gram-positive bacteria and more diverse Bacillus species. Virology 2018, 518, 136–142. [Google Scholar] [CrossRef]
- Kan, S.; Fornelos, N.; Schuch, R.; Fischetti, V.A. Identification of a Ligand on the Wip1 Bacteriophage Highly Specific for a Receptor on Bacillus anthracis. J. Bacteriol. 2013, 195, 4355–4364. [Google Scholar] [CrossRef] [Green Version]
- Sozhamannan, S.; McKinstry, M.; Lentz, S.M.; Jalasvuori, M.; McAfee, F.; Smith, A.; Dabbs, J.; Ackermann, H.-W.; Bamford, J.K.H.; Mateczun, A.; et al. Molecular Characterization of a variant of Bacillus anthracis-specific phage AP50 with improved bacteriolytic activity. Appl. Environ. Microbiol. 2008, 74, 6792–6796. [Google Scholar] [CrossRef] [Green Version]
- Gillis, A.; Mahillon, J. Phages Preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, Present and Future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [Green Version]
- Ravantti, J.J.; Gaidelyte, A.; Bamford, D.H.; Bamford, J.K. Comparative analysis of bacterial viruses Bam35, infecting a gram-positive host, and PRD1, infecting gram-negative hosts, demonstrates a viral lineage. Virology 2003, 313, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Laurinmäki, P.; Huiskonen, J.; Bamford, D.; Butcher, S. Membrane Proteins Modulate the Bilayer Curvature in the Bacterial Virus Bam35. Structure 2005, 13, 1819–1828. [Google Scholar] [CrossRef] [Green Version]
- Otero, M.B.; Villar, L.; Salas, M.; Redrejo-Rodríguez, M. Disclosing early steps of protein-primed genome replication of the Gram-positive tectivirus Bam35. Nucleic Acids Res. 2016, 44, 9733–9744. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.-W.; Roy, R.; Martin, M.; Murthy, M.R.V.; Smirnoff, W.A. Partial characterization of a cubic Bacillus phage. Can. J. Microbiol. 1978, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Palma, L.; Muñoz, D.; Berry, C.; Murillo, J.; Caballero, P. Bacillus thuringiensis Toxins: An Overview of Their Biocidal Activity. Toxins 2014, 6, 3296–3325. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, V. From microbial sprays to insect-resistant transgenic plants: History of the biospesticide Bacillus thuringiensis. A review. Agron. Sustain. Dev. 2010, 31, 217–231. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Prevalence, Genetic Diversity and Host Range of Tectiviruses among Members of the Bacillus cereus Group. Appl. Environ. Microbiol. 2014, 80, 4138–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornelos, N.; Butala, M.; Hodnik, V.; Anderluh, G.; Bamford, J.K.; Salas, M. Bacteriophage GIL01 gp7 interacts with host LexA repressor to enhance DNA binding and inhibit RecA-mediated auto-cleavage. Nucleic Acids Res. 2015, 43, 7315–7329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berjón-Otero, M.; Lechuga, A.; Mehla, J.; Uetz, P.; Salas, M.; Redrejo-Rodríguez, M. Bam35 Tectivirus Intraviral Interaction Map Unveils New Function and Localization of Phage ORFan Proteins. J. Virol. 2017, 91, e00870-17. [Google Scholar] [CrossRef] [Green Version]
- Laurinavičius, S.; Käkelä, R.; Somerharju, P.; Bamford, D.H. Phospholipid molecular species profiles of tectiviruses infecting Gram-negative and Gram-positive hosts. Virology 2004, 322, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Verheust, C.; Fornelos, N.; Mahillon, J.; Verheust, C. The Bacillus thuringiensis phage GIL01 encodes two enzymes with peptidoglycan hydrolase activity. FEMS Microbiol. Lett. 2004, 237, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Gaidelytė, A.; Cvirkaitė-Krupovic, V.; Daugelavicius, R.; Bamford, J.K.H.; Bamford, D.H. The Entry Mechanism of Membrane-Containing Phage Bam35 Infecting Bacillus thuringiensis. J. Bacteriol. 2006, 188, 5925–5934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahn, A.M.; Daugelavicius, R.; Bamford, D.H. Sequential model of phage PRD1 DNA delivery: Active involvement of the viral membrane. Mol. Microbiol. 2002, 46, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Salas, M.; de Vega, M. Protein-Primed Replication of Bacteriophage Φ29 DNA. Enzymes 2016, 39, 137–167. [Google Scholar] [CrossRef]
- Otero, M.B.; Villar, L.; de Vega, M.; Salas, M.; Redrejo-Rodríguez, M. DNA polymerase from temperate phage Bam35 is endowed with processive polymerization and abasic sites translesion synthesis capacity. Proc. Natl. Acad. Sci. USA 2015, 112, E3476–E3484. [Google Scholar] [CrossRef] [Green Version]
- Verheust, C.; Jensen, G.; Mahillon, J. pGIL01, a linear tectiviral plasmid prophage originating from Bacillus thuringiensis serovar israelensis. Microbiology 2003, 149, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornelos, N.; Bamford, J.K.H.; Mahillon, J. Phage-Borne Factors and Host LexA Regulate the Lytic Switch in Phage GIL01. J. Bacteriol. 2011, 193, 6008–6019. [Google Scholar] [CrossRef] [Green Version]
- Caveney, N.; Pavlin, A.; Caballero, G.; Bahun, M.; Hodnik, V.; De Castro, L.; Fornelos, N.; Butala, M.; Strynadka, N.C. Structural Insights into Bacteriophage GIL01 gp7 Inhibition of Host LexA Repressor. Structure 2019, 27, 1094–1102. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Influence of Lysogeny of Tectiviruses GIL01 and GIL16 on Bacillus thuringiensis Growth, Biofilm Formation, and Swarming Motility. Appl. Environ. Microbiol. 2014, 80, 7620–7630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornelos, N.; Browning, D.F.; Pavlin, A.; Podlesek, Z.; Hodnik, V.; Salas, M.; Butala, M. Lytic gene expression in the temperate bacteriophage GIL01 is activated by a phage-encoded LexA homologue. Nucleic Acids Res. 2018, 46, 9432–9443. [Google Scholar] [CrossRef]
- Gaidelytė, A.; Jaatinen, S.T.; Daugelavičius, R.; Bamford, J.K.H.; Bamford, D.H. The Linear Double-Stranded DNA of Phage Bam35 Enters Lysogenic Host Cells, but the Late Phage Functions Are Suppressed. J. Bacteriol. 2005, 187, 3521–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, P.J.; Federspiel, J.D.; Sheng, X.; Cristea, I.M. Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol. Syst. Biol. 2017, 13, 922. [Google Scholar] [CrossRef]
- Fields, S.; Song, O.-K. A novel genetic system to detect protein–protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef]
- Bartel, P.L.; Roecklein, J.A.; Sengupta, D.; Fields, S. A protein linkage map of Escherichia coli bacteriophage T7. Nat. Genet. 1996, 12, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Lecoutere, E.; Chibeu, A.; Hertveldt, K.; Volckaert, G.; Lavigne, R. A procedure for systematic identification of bacteriophage–host interactions of P. aeruginosa phages. Virology 2009, 387, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasche, S.; Wuchty, S.; Rajagopala, S.; Uetz, P. The Protein Interaction Network of Bacteriophage Lambda with Its Host, Escherichia coli. J. Virol. 2013, 87, 12745–12755. [Google Scholar] [CrossRef] [Green Version]
- Wagemans, J.; Blasdel, B.; Bossche, A.V.D.; Uytterhoeven, B.; De Smet, J.; Paeshuyse, J.; Cenens, W.; Aertsen, A.; Uetz, P.; Delattre, A.-S.; et al. Functional elucidation of antibacterial phage ORFans targeting Pseudomonas aeruginosa. Cell. Microbiol. 2014, 16, 1822–1835. [Google Scholar] [CrossRef]
- Mariano, R.; Wuchty, S.; Vizoso-Pinto, M.G.; Hauser, R.; Uetz, P. The interactome of Streptococcus pneumoniae and its bacteriophages show highly specific patterns of interactions among bacteria and their phages. Sci. Rep. 2016, 6, 24597. [Google Scholar] [CrossRef] [Green Version]
- Mehla, J.; Dedrick, R.M.; Caufield, J.H.; Wagemans, J.; Sakhawalkar, N.; Johnson, A.; Hatfull, G.F.; Uetz, P. Virus-host protein-protein interactions of mycobacteriophage Giles. Sci. Rep. 2017, 7, 16514. [Google Scholar] [CrossRef] [Green Version]
- Häuser, R.; Blasche, S.; Dokland, T.; Haggård-Ljungquist, E.; von Brunn, A.; Salas, M.; Casjens, S.; Molineux, I.; Uetz, P. Bacteriophage Protein–Protein Interactions. Adv. Virus Res. 2012, 83, 219–298. [Google Scholar] [CrossRef] [Green Version]
- Bossche, A.V.D.; Ceyssens, P.-J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.-S.; Cenens, W.; Aertsen, A.; et al. Systematic Identification of Hypothetical Bacteriophage Proteins Targeting Key Protein Complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef] [PubMed]
- Mariano, R.; Khuri, S.; Uetz, P.; Wuchty, S. Local Action with Global Impact: Highly Similar Infection Patterns of Human Viruses and Bacteriophages. mSystems 2016, 1, e00030-15. [Google Scholar] [CrossRef] [Green Version]
- Yachie, N.; Petsalaki, E.; Mellor, J.C.; Weile, J.; Jacob, Y.; Verby, M.; Ozturk, S.B.; Li, S.; Cote, A.G.; Mosca, R.; et al. Pooled-matrix protein interaction screens using Barcode Fusion Genetics. Mol. Syst. Biol. 2016, 12, 863. [Google Scholar] [CrossRef]
- Trigg, S.; Garza, R.M.; MacWilliams, A.; Nery, J.R.; Bartlett, A.; Castanon, R.; Goubil, A.; Feeney, J.; O’Malley, R.; Huang, S.-S.C.; et al. CrY2H-seq: A massively multiplexed assay for deep-coverage interactome mapping. Nat. Methods 2017, 14, 819–825. [Google Scholar] [CrossRef]
- Yang, J.-S.; Garriga-Canut, M.; Link, N.; Carolis, C.; Broadbent, K.; Beltran-Sastre, V.; Serrano, L.; Maurer, S.P. rec-YnH enables simultaneous many-by-many detection of direct protein–protein and protein–RNA interactions. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.D.; Wan, J.; Ford, R.; Gong, Y.; Fung, P.; Nahal, H.; Wang, P.W.; Desveaux, D.; Guttman, D.S. Quantitative Interactor Screening with next-generation Sequencing (QIS-Seq) identifies Arabidopsis thaliana MLO2 as a target of the Pseudomonas syringae type III effector HopZ2. BMC Genom. 2012, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erffelinck, M.-L.; Ribeiro, B.; Perassolo, M.; Pauwels, L.; Pollier, J.; Storme, V.; Goossens, A. A user-friendly platform for yeast two-hybrid library screening using next generation sequencing. PLoS ONE 2018, 13, e0201270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehla, J.; Caufield, J.; Sakhawalkar, N.; Uetz, P. A Comparison of Two-Hybrid Approaches for Detecting Protein–Protein Interactions. Methods Enzymol. 2017, 586, 333–358. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, T.; Robert-Baudouy, J. Bacterial aminopeptidases: Properties and functions. FEMS Microbiol. Rev. 1996, 18, 319–344. [Google Scholar] [CrossRef]
- Lechuga, A.; Kazlauskas, D.; Salas, M.; Redrejo-Rodríguez, M. Unlimited Cooperativity of Betatectivirus SSB, a Novel DNA Binding Protein Related to an Atypical Group of SSBs from Protein-Primed Replicating Bacterial Viruses. Front. Microbiol. 2021, 12, 699140. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Yamamoto, N.; Zweifel, A.E.; Nakamichi, T.; Huang, H.-K.; Mendez-Rios, J.D.; Franca-Koh, J.; Boorgula, M.P.; Fujita, K.; Suzuki, K.-I.; et al. The Escherichia coli K-12 ORFeome: A resource for comparative molecular microbiology. BMC Genom. 2010, 11, 470. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.S.; Schaefer-Ramadan, S.; Al-Thani, N.; Ahmed, I.; Mohamoud, Y.A.; Malek, J.A. High-resolution protein–protein interaction mapping using all-versus-all sequencing (AVA-Seq). J. Biol. Chem. 2019, 294, 11549–11558. [Google Scholar] [CrossRef]
- Lechuga, A.; Lood, C.; Salas, M.; van Noort, V.; Lavigne, R.; Redrejo-Rodríguez, M. Completed Genomic Sequence of Bacillus thuringiensis HER1410 Reveals a Cry-Containing Chromosome, Two Megaplasmids, and an Integrative Plasmidial Prophage. G3 Genes Genomes Genet. 2020, 10, 2927–2939. [Google Scholar] [CrossRef]
- Maier, R.H.; Brandner, C.J.; Hintner, H.; Bauer, J.W.; Önder, K. Construction of a reading frame–independent yeast two-hybrid vector system for site-specific recombinational cloning and protein interaction screening. BioTechniques 2008, 45, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Lei, Y.; Zhou, M.; Yao, Q.; Han, Y.; Wu, X.; Zhong, W.; Zhu, C.; Xu, W.; Tao, R.; et al. Development and application of a recombination-based library versus library high- throughput yeast two-hybrid (RLL-Y2H) screening system. Nucleic Acids Res. 2017, 46, e17. [Google Scholar] [CrossRef] [Green Version]
- Caufield, J.H.; Sakhawalkar, N.; Uetz, P. A comparison and optimization of yeast two-hybrid systems. Methods 2012, 58, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Stellberger, T.; Häuser, R.; Baiker, A.; Pothineni, V.R.; Haas, J.; Uetz, P. Improving the yeast two-hybrid system with permutated fusions proteins: The Varicella Zoster Virus interactome. Proteome Sci. 2010, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Rajagopala, S.; Sikorski, P.; Kumar, A.; Mosca, R.; Vlasblom, J.; Arnold, R.; Franca-Koh, J.; Pakala, S.; Phanse, S.; Ceol, A.; et al. The binary protein-protein interaction landscape of Escherichia coli. Nat. Biotechnol. 2014, 32, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Wuchty, S.; Rajagopala, S.V.; Blazie, S.M.; Parrish, J.R.; Khuri, S.; Finley, R.; Uetz, P. The Protein Interactome of Streptococcus pneumoniae and Bacterial Meta-interactomes Improve Function Predictions. mSystems 2017, 2, e00019-17. [Google Scholar] [CrossRef] [Green Version]
- Suter, B.; Zhang, X.; Pesce, C.G.; Mendelsohn, A.R.; Dinesh-Kumar, S.P.; Mao, J.-H. Next-Generation Sequencing for Binary Protein–Protein Interactions. Front. Genet. 2015, 6, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef] [Green Version]
- Weimann, M.; Grossmann, A.; Woodsmith, J.; Özkan, Z.; Birth, P.; Meierhofer, D.; Benlasfer, N.; Valovka, T.; Timmermann, B.; Wanker, E.; et al. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat. Methods 2013, 10, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Fayad, N.; Makart, L.; Bolotin, A.; Sorokine, A.; Kallassy, M.; Mahillon, J. Role of plasmid plasticity and mobile genetic elements in the entomopathogen Bacillus thuringiensis serovar israelensis. FEMS Microbiol. Rev. 2018, 42, 829–856. [Google Scholar] [CrossRef]
- Pfeifer, E.; Sousa, J.A.M.D.; Touchon, M.; Rocha, E.P.C. Bacteria have numerous distinctive groups of phage–plasmids with conserved phage and variable plasmid gene repertoires. Nucleic Acids Res. 2021, 49, 2655–2673. [Google Scholar] [CrossRef] [PubMed]
- Pelchovich, G.; Omer-Bendori, S.; Gophna, U. Menaquinone and Iron Are Essential for Complex Colony Development in Bacillus subtilis. PLoS ONE 2013, 8, e79488. [Google Scholar] [CrossRef] [Green Version]
- Madeira, J.-P.; Omer, H.; Alpha-Bazin, B.; Armengaud, J.; Duport, C. Deciphering the interactions between the Bacillus cereus linear plasmid, pBClin15, and its host by high-throughput comparative proteomics. J. Proteom. 2016, 146, 25–33. [Google Scholar] [CrossRef]
- Poranen, M.; Ravantti, J.J.; Grahn, A.M.; Gupta, R.; Auvinen, P.; Bamford, D.H. Global Changes in Cellular Gene Expression during Bacteriophage PRD1 Infection. J. Virol. 2006, 80, 8081–8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojardín, L.; Salas, M. Global Transcriptional Analysis of Virus-Host Interactions between Phage ϕ29 and Bacillus subtilis. J. Virol. 2016, 90, 9293–9304. [Google Scholar] [CrossRef] [Green Version]
- Leskinen, K.; Blasdel, B.G.; Lavigne, R.; Skurnik, M. RNA-Sequencing Reveals the Progression of Phage-Host Interactions between φR1-37 and Yersinia enterocolitica. Viruses 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, C.; Danis-Wlodarczyk, K.; Blasdel, B.G.; Bin Jang, H.; Vandenheuvel, D.; Briers, Y.; Noben, J.; van Noort, V.; Drulis-Kawa, Z.; Lavigne, R. Integrative omics analysis of Pseudomonas aeruginosa virus PA5oct highlights the molecular complexity of jumbo phages. Environ. Microbiol. 2020, 22, 2165–2181. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Brombacher, E.; Dorel, C.; Zehnder, A.J.B.; Landini, P. The curli biosynthesis regulator CsgD co-ordinates the expression of both positive and negative determinants for biofilm formation in Escherichia coli. Microbiology 2003, 149, 2847–2857. [Google Scholar] [CrossRef] [Green Version]
- Mattila, S.; Oksanen, H.M.; Bamford, J.K.H. Probing protein interactions in the membrane-containing virus PRD1. J. Gen. Virol. 2015, 96, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Rydman, P.S.; Bamford, D.H. Identification and Mutational Analysis of Bacteriophage PRD1 Holin Protein P35. J. Bacteriol. 2003, 185, 3795–3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänninen, A.-L.; Bamford, D.H.; Bamford, J.K. Assembly of Membrane-Containing Bacteriophage PRD1 Is Dependent on GroEL and GroES. Virology 1997, 227, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Clair, G.; Roussi, S.; Armengaud, J.; Duport, C. Expanding the Known Repertoire of Virulence Factors Produced by Bacillus cereus through Early Secretome Profiling in Three Redox Conditions. Mol. Cell. Proteom. 2010, 9, 1486–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamitori, S.; Yoshida, H. Structure-Function Relationship of Bacterial SH3 Domains. In SH Domains: Structure, Mechanisms and Applications; Kurochkina, N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 71–89. [Google Scholar] [CrossRef]
- Jalasvuori, M.; Palmu, S.; Gillis, A.; Kokko, H.; Mahillon, J.; Bamford, J.K.; Fornelos, N. Identification of five novel tectiviruses in Bacillus strains: Analysis of a highly variable region generating genetic diversity. Res. Microbiol. 2013, 164, 118–126. [Google Scholar] [CrossRef]
- Charlier, D.; Gh, G.H.; Kholti, A.; Gigot, D.; Piérard, A.; Glansdorff, N. carP, Involved in Pyrimidine Regulation of the Escherichia coli Carbamoylphosphate Synthetase Operon Encodes a Sequence-specific DNA-binding Protein Identical to XerB and PepA, also Required for Resolution of ColEl Multimers. J. Mol. Biol. 1995, 250, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Moses, S.; Sinner, T.; Zaprasis, A.; Stöveken, N.; Hoffmann, T.; Belitsky, B.R.; Sonenshein, A.L.; Bremer, E. Proline Utilization by Bacillus subtilis: Uptake and Catabolism. J. Bacteriol. 2012, 194, 745–758. [Google Scholar] [CrossRef] [Green Version]
- James, P.; Halladay, J.; Craig, E.A. Genomic Libraries and a Host Strain Designed for Highly Efficient Two-Hybrid Selection in Yeast. Genetics 1996, 144, 1425–1436. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Precipitation of DNA with Ethanol. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Clarke, L.; Carbon, J. A colony bank containing synthetic CoI EI hybrid plasmids representative of the entire E. coli genome. Cell 1976, 9, 91–99. [Google Scholar] [CrossRef]
- Elsaesser, R.; Paysan, J. Liquid gel amplification of complex plasmid libraries. BioTechniques 2004, 37, 200–202. [Google Scholar] [CrossRef]
- Kriegler, M. Gene Transfer and Expression: A Laboratory Manual; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Benatuil, L.; Perez, J.M.; Belk, J.; Hsieh, C.-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010, 23, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Mehla, J.; Caufield, J.H.; Uetz, P. Mapping Protein–Protein Interactions Using Yeast Two-Hybrid Assays. Cold Spring Harb. Protoc. 2015, 2015, pdb-prot086157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seqtk. Available online: https://github.com/lh3/seqtk (accessed on 25 October 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2006, 35, D5–D12. [Google Scholar] [CrossRef]
- Li, H. BWA-MEM. Available online: https://github.com/lh3/bwa (accessed on 12 October 2021).
- Li, H.; Handsaker, R.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.r-project.org/ (accessed on 12 October 2021).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment | Number of Interactions | Read Counts (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pBC | pBN | Total | pBC | pBN | Total | ||||||
| Category | Range | pPC Library | pPN Library | pPC Library | pPN Library | pPC Library | pPN Library | pPC Library | pPN Library | ||
| A | 10–100% | 93 | 43 | 67 | 27 | 230 | 77.89 | 72.61 | 73.75 | 81.09 | 75.93 |
| B | 0.25–10% | 262 | 165 | 391 | 95 | 913 | 20.76 | 24.34 | 24.52 | 15.76 | 22.03 |
| C | 0–0.25% | 738 | 794 | 990 | 812 | 3334 | 1.35 | 3.05 | 1.73 | 3.15 | 2.04 |
| Total | 1093 | 1002 | 1448 | 934 | 4477 | 100 | 100 | 100 | 100 | 100 | |
| Combination | Total | A + B Categories | No “Sticky” Prey | No Empty | No Duplicates |
|---|---|---|---|---|---|
| pBC–pPC Library | 1093 | 355 | 31 | 25 | 15 |
| pBC–pPN Library | 1002 | 208 | 24 | 15 | 14 |
| pBN–pPC Library | 1448 | 458 | 119 | 119 | 106 |
| pBN–pPN Library | 934 | 122 | 54 | 52 | 47 |
| Total | 4477 | 1143 | 228 | 211 | 182 |
| Bait | Prey | Max. 3AT (mM) | Max. 3AT (mM) (Emptybait_BtORF) | Max. 3AT (mM) | Interaction | Bt Protein Coverage |
|---|---|---|---|---|---|---|
| (B35ORF) | (BtORF) | (B35ORF_Emptyprey) | (B35ORF_BtORF) | |||
| pBC_06 | pPC_pepA(24805) | 0.1 | 50 | 0.1 | N/A | 0.33 |
| pBC_10 | pPC_dapF(25140) | 50 | 10 | 10 | N/A | 0.44 |
| pBC_25 | pPC_hutI(18010) | 0.025 | 50 | No growth | No | 0.43 |
| pBC_25 | pPC_pepA(24805) | 0.025 | 50 | 0 | N/A | 0.33 |
| pBC_26 | pPN_pepD(12045) | No growth | 25 | No growth | No | 0.37 |
| pBC_31 | pPC_hutI(18010) | 0 | 50 | 0 | N/A | 0.43 |
| pBC_31 | pPC_pepA(24805) | 0 | 50 | 0 | N/A | 0.33 |
| pBC_31 | pPC_tyrS(25855) | 0 | 10 | 0 | N/A | 0.15 |
| pBN_03 | pPC_pbpX(02235) | No growth | 0.1 | No growth | No | 0.44 |
| pBN_06 | pPC_pepA(24805) | No growth | 0.1 | No growth | No | 0.33 |
| pBN_08 | pPN_lexA(18215) | No growth | 0.1 | No growth | No | 0.42 |
| pBN_11 | pPC_pbpX(02235) | No growth | 0.1 | No growth | No | 0.44 |
| pBN_15 | pPC_iap(27190) | No growth | 0.1 | 25 | Yes | 0.76 |
| pBN_15 | pPN_(30985) | No growth | 0.1 | No growth | No | 0.18 |
| pBN_16 | pPC_iap(27190) | No growth | 0.1 | 25 | Yes | 0.76 |
| pBN_16 | pPN_(30985) | No growth | 0.1 | No growth | No | 0.18 |
| pBN_19 | pPC_iap(27190) | No growth | 0.1 | 50 | Yes | 0.76 |
| pBN_19t | pPC_ompR(22050) | 50 | 10 | 100 | Yes | 0.99 |
| pBN_19t | pPN_menF(24455) | 50 | 0.1 | 50 | N/A | 0.31 |
| pBN_20 | pPC_purL(01870) | No growth | 0.1 | No growth | No | 0.24 |
| pBN_22 | pPC_yadS(28175) | 0 | 0.1 | 50 | Yes | 0.74 |
| pBN_22 | pPN_(30985) | No growth | 0.1 | 0 | N/A | 0.18 |
| pBN_24 | pPN_usp(26960) | 0.025 | 0.1 | 0.1 | N/A | 0.54 |
| pBN_25 | pPC_hutI(18010) | No growth | 0.1 | No growth | No | 0.43 |
| pBN_25 | pPC_pepA(24805) | No growth | 0.1 | No growth | No | 0.33 |
| pBN_25t | pPC_pepA(24805) | 25 | 0.1 | 25 | N/A | 0.33 |
| pBN_25t | pPN_pepA(24805) | 25 | 0.1 | 25 | N/A | 0.33 |
| pBN_26 | pPC_(16200) | No growth | 0.1 | 3 | Yes | 0.89 |
| pBN_26 | pPN_(30985) | No growth | 0.1 | No growth | No | 0.18 |
| pBN_27 | pPC_iap(27190) | 0.025 | 0.1 | 50 | Yes | 0.76 |
| pBN_27 | pPN_(30985) | No growth | 0.1 | No growth | No | 0.18 |
| pBN_27t | pPN_usp(26960) | 3 | 0.1 | 0.1 | N/A | 0.54 |
| pBN_31 | pPC_hutI(18010) | 0.1 | 0.1 | No growth | No | 0.43 |
| B35 Protein (Function) | Bt COG b Interactions | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B | C | D | E | F | G | H | I | J | K | L | M | NO | O | P | Q | S | T | U | V | Total | ||
| Gene regulation and Genome replication | 1 (DNA binding/phage cycle regulator *) | 1 | 1 | |||||||||||||||||||
| 2 (SSB) | 1 | 1 | 2 | |||||||||||||||||||
| 3 (Unknown) | 1 | 1 | 2 | |||||||||||||||||||
| 4 (TP) | 1 | 1 | 1 | 3 | ||||||||||||||||||
| 6 (Cycle regulator) | 1 | 2 | 1 | 1 | 1 | 1 | 7 | |||||||||||||||
| 7 (Cycle regulator) | 1 | 1 | ||||||||||||||||||||
| 8 (Unknown) | 1 | 1 | ||||||||||||||||||||
| Virion structure and DNA packaging | 9 (Unknown) | 1 | 1 | 1 | 3 | |||||||||||||||||
| 10 (DNA packaging *) | 1 | 2 | 1 | 4 | ||||||||||||||||||
| 11 (Assembly protein *) | 1 | 1 | 2 | |||||||||||||||||||
| 12 (Unknown) | 1 | 1 | ||||||||||||||||||||
| 13 (Unknown) | 1 | 3 | 1 | 1 | 1 | 2 | 1 | 10 | ||||||||||||||
| 14 (DNA packaging/ATPase *) | 1 | 1 | ||||||||||||||||||||
| 15 (Special vertex *) | 3 | 1 | 2 | 1 | 1 | 1 | 1 | 10 | ||||||||||||||
| 16 (DNA packaging *) | 1 | 1 | 1 | 3 | ||||||||||||||||||
| 17 (Coat minor capsid protein *) | 1 | 1 | 1 | 1 | 2 | 6 | ||||||||||||||||
| 19 (DNA packaging *) | 3 | 1 | 1 | 1 | 3 | 1 | 10 | |||||||||||||||
| 19t (DNA packaging *) | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||||||||||
| 20 (Stabilizer of spike *) | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 9 | ||||||||||||||
| 20t (Stabilizer of spike *) | 1 | 1 | 1 | 3 | ||||||||||||||||||
| 21 (Unknown) | 1 | 1 | 1 | 3 | ||||||||||||||||||
| 22 (Special vertex *) | 1 | 1 | 1 | 2 | 3 | 8 | ||||||||||||||||
| 23 (Unknown) | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 9 | ||||||||||||||
| 24 (Penton *) | 1 | 1 | 1 | 1 | 1 | 1 | 6 | |||||||||||||||
| 25 (Membrane structural component) | 1 | 3 | 1 | 1 | 1 | 2 | 2 | 11 | ||||||||||||||
| 25t (Membrane structural component) | 2 | 1 | 1 | 1 | 1 | 6 | ||||||||||||||||
| 26 (Transglycosylase and integral membrane scaffolding *) | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 5 | 2 | 17 | ||||||||||||
| 26t (Transglycosylase and integral membrane scaffolding *) | 1 | 2 | 3 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 15 | |||||||||||
| 27 (Unknown) | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 10 | ||||||||||||||
| 27t (Unknown) | 1 | 1 | ||||||||||||||||||||
| Host recognition and lysis | 28 (Spike *) | 1 | 1 | 2 | ||||||||||||||||||
| 29 (Spike *) | 1 | 1 | 1 | 1 | 4 | |||||||||||||||||
| 30 (Transglycosylase) | 1 | 1 | 2 | |||||||||||||||||||
| 31 (Unknown) | 1 | 1 | 1 | 1 | 4 | |||||||||||||||||
| Total | 1 | 20 | 5 | 24 | 7 | 16 | 6 | 4 | 6 | 2 | 5 | 6 | 5 | 6 | 14 | 7 | 31 | 9 | 1 | 7 | 182 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lechuga, A.; Lood, C.; Berjón-Otero, M.; del Prado, A.; Wagemans, J.; van Noort, V.; Lavigne, R.; Salas, M.; Redrejo-Rodríguez, M. Unraveling Protein Interactions between the Temperate Virus Bam35 and Its Bacillus Host Using an Integrative Yeast Two Hybrid–High Throughput Sequencing Approach. Int. J. Mol. Sci. 2021, 22, 11105. https://doi.org/10.3390/ijms222011105
Lechuga A, Lood C, Berjón-Otero M, del Prado A, Wagemans J, van Noort V, Lavigne R, Salas M, Redrejo-Rodríguez M. Unraveling Protein Interactions between the Temperate Virus Bam35 and Its Bacillus Host Using an Integrative Yeast Two Hybrid–High Throughput Sequencing Approach. International Journal of Molecular Sciences. 2021; 22(20):11105. https://doi.org/10.3390/ijms222011105
Chicago/Turabian StyleLechuga, Ana, Cédric Lood, Mónica Berjón-Otero, Alicia del Prado, Jeroen Wagemans, Vera van Noort, Rob Lavigne, Margarita Salas, and Modesto Redrejo-Rodríguez. 2021. "Unraveling Protein Interactions between the Temperate Virus Bam35 and Its Bacillus Host Using an Integrative Yeast Two Hybrid–High Throughput Sequencing Approach" International Journal of Molecular Sciences 22, no. 20: 11105. https://doi.org/10.3390/ijms222011105
APA StyleLechuga, A., Lood, C., Berjón-Otero, M., del Prado, A., Wagemans, J., van Noort, V., Lavigne, R., Salas, M., & Redrejo-Rodríguez, M. (2021). Unraveling Protein Interactions between the Temperate Virus Bam35 and Its Bacillus Host Using an Integrative Yeast Two Hybrid–High Throughput Sequencing Approach. International Journal of Molecular Sciences, 22(20), 11105. https://doi.org/10.3390/ijms222011105