
Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis


Abstract
:1. Introduction
2. Results
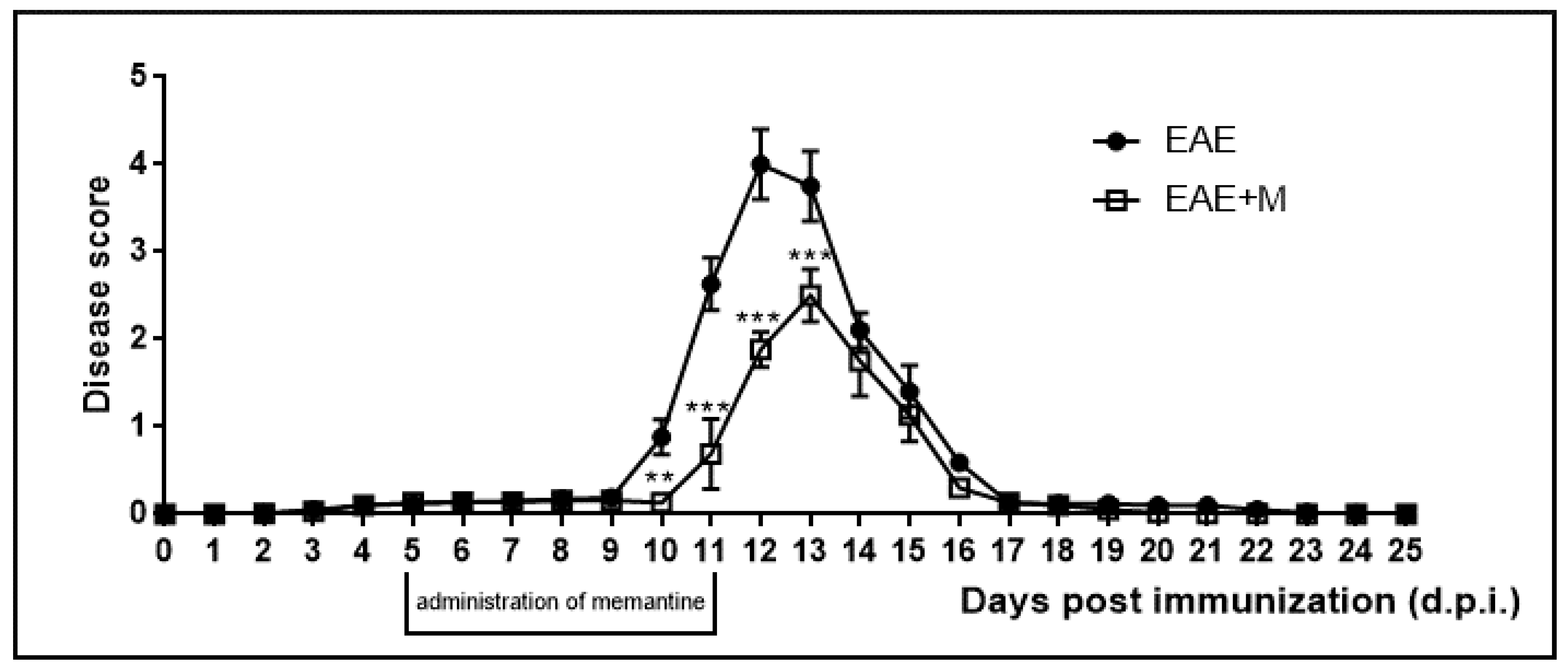
2.1. The Effect of Memantine on the Course of EAE
2.2. The Effect of NMDA Receptor Antagonists on the Level of Oxidative Stress Markers
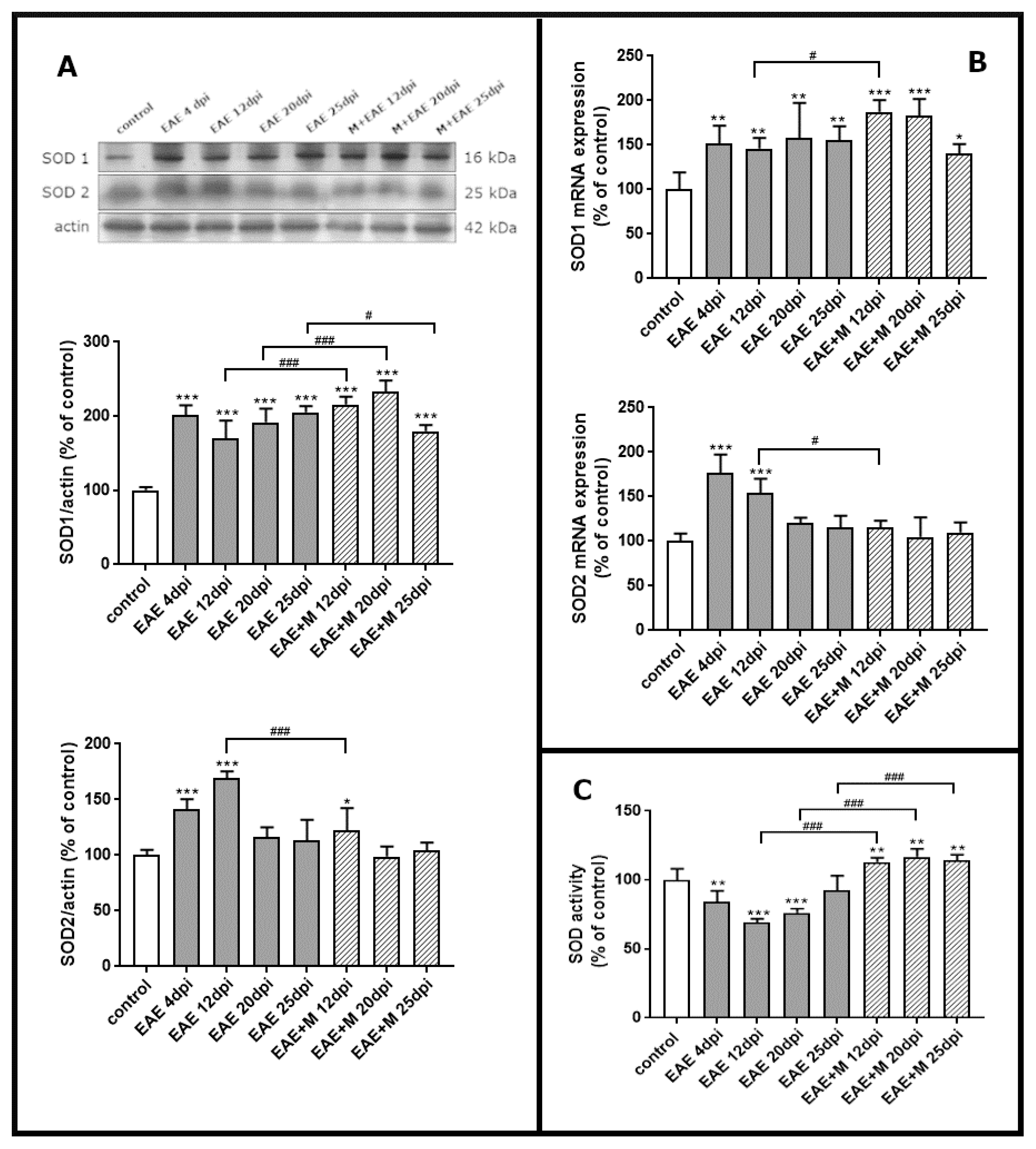
2.3. Memantine-Induced Changes in the Expression of SOD Enzymes
2.4. Memantine-Dependent Changes in SOD’s Activity
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Experimental Groups and Tissue Processing
4.3. Measurement of Lipid Peroxidation
4.4. Measurement of the Level of Sulfhydryl Groups
4.5. Western Blot Analysis
4.6. Determination of SOD’s Expression by Quantitative Real Time PCR
4.7. Measurement of SOD’s Activity
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C.; Canadian Collaborative Study Group. Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol. 2006, 5, 932–936. [Google Scholar] [CrossRef]
- Groom, A.J.; Smith, T.; Turski, L. Multiple sclerosis and glutamate. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Dittel, B.N. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 2005, 81, 374–389. [Google Scholar] [CrossRef]
- Cuzner, M.L.; Hayes, G.M.; Newcombe, J.; Wooroofe, M.N. The nature of inflammatory components during demyelination in multiple sclerosis. J. Neuroimmunol. 1988, 20, 203–209. [Google Scholar] [CrossRef]
- Van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef]
- Gonsette, R.E. Neurodegeneration in multiple sclerosis: The role of oxidative stress and excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Laurie, D.J.; Berthele, A.; Sommer, B.; Tölle, T.R.; Gebicke-Härter, P.-J.; Van Calker, D.; Boddeke, H.W.G.M. Expression and Signaling of Group I Metabotropic Glutamate Receptors in Astrocytes and Microglia. J. Neurochem. 1999, 72, 1671–1680. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Li, F. Driving Cellular Plasticity and Survival Through the Signal Transduction Pathways of Metabotropic Glutamate Receptors. Curr. Neurovasc. Res. 2005, 2, 425–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidoryk-Wegrzynowicz, M.; Struzynska, L. Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 8404. [Google Scholar] [CrossRef]
- Choi, D.W. Calcium and Excitotoxic Neuronal Injury. Ann. N. Y. Acad. Sci. 1994, 747, 162–171. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Bolton, C. Modulation of Blood-Brain Barrier Dysfunction and Neurological Deficits during Acute Experimental Allergic Encephalomyelitis by theN-Methyl-d-aspartate Receptor Antagonist Memantine. J. Pharmacol. Exp. Ther. 2002, 302, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Role of LOX/COX pathways in 3-nitropropionic acid-induced Huntington’s Disease-like symptoms in rats: Protective effect of licofelone. Br. J. Pharmacol. 2011, 164, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Lipton, S.A. NMDA receptor activity regulates transcription of antioxidant pathways. Nat. Neurosci. 2008, 11, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Fiskum, G. Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 2004, 90, 1281–1289. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Pellergini, J.W.; Aggarwal, S.K.; Lei, S.Z.; Warach, S.; Jensen, F.E.; Lipton, S.A. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: Therapeutic advantage against NMDA receptor-mediated neurotoxicity. J. Neurosci. 1992, 12, 4427–4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, E.K.; Levine, J.D. Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 2004, 20, 2896–2902. [Google Scholar] [CrossRef]
- Marini, A.M.; Ueda, Y.; June, C.H. Intracellular Survival Pathways against Glutamate Receptor Agonist Excitotoxicity in Cultured Neurons: Intracellular Calcium Responses. Ann. N. Y. Acad. Sci. 1999, 890, 421–437. [Google Scholar] [CrossRef]
- Kean, R.B.; Spitsin, S.V.; Mikheeva, T.; Scott, G.S.; Hooper, D.C. The Peroxynitrite Scavenger Uric Acid Prevents Inflammatory Cell Invasion into the Central Nervous System in Experimental Allergic Encephalomyelitis through Maintenance of Blood-Central Nervous System Barrier Integrity. J. Immunol. 2000, 165, 6511–6518. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C.; Scott, G.S.; Zborek, A.; Mikheeva, T.; Kean, R.B.; Koprowski, H.; Spitsin, S.V. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB J. 2000, 14, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Paul, C. Glutamate Receptors in Neuroinflammatory Demyelinating Disease. Mediat. Inflamm. 2006, 2006, 1–12. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; Van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.S.-H.; Zhao, M.-L.; Brosnan, C.F.; Lee, S.C. Expression of Inducible Nitric Oxide Synthase and Nitrotyrosine in Multiple Sclerosis Lesions. Am. J. Pathol. 2001, 158, 2057–2066. [Google Scholar] [CrossRef] [Green Version]
- LeVine, S.M. The role of reactive oxygen species in the pathogenesis of multiple sclerosis. Med. Hypotheses 1992, 39, 271–274. [Google Scholar] [CrossRef]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Strużyńska, L. Modulation of Neurological Deficits and Expression of Glutamate Receptors during Experimental Autoimmune Encephalomyelitis after Treatment with Selected Antagonists of Glutamate Receptors. Biomed Res. Int. 2013, 2013, 186068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, G.; Dabrowska-Bouta, B.; Chalimoniuk, M.; Struzyńska, L. Effects of antagonists of glutamate receptors on pro-inflammatory cytokines in the brain cortex of rats subjected to experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 261, 67–76. [Google Scholar] [CrossRef]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Salińska, E.; Strużyńska, L. Modulation of Glutamate Transport and Receptor Binding by Glutamate Receptor Antagonists in EAE Rat Brain. PLoS ONE 2014, 9, 113954. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, K.; Chuchmacz, J.; Wójtowicz, P.; Szymański, P.J. Memantine in neurological disorders—Schizophrenia and depression. J. Mol. Med. 2021, 99, 327–334. [Google Scholar] [CrossRef]
- Chen, H.S.; Lipton, S.A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006, 97, 1611–1626. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-S.V.; Lipton, S.A. Pharmacological implications of two distinct mechanisms of interaction of memantine with N-me-thyl-D-aspartate-gated channels. J. Pharmacol. Exp. Ther. 2005, 314, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Volbracht, C.; Van Beek, J.; Zhu, C.; Blomgren, K.; Leist, M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur. J. Neurosci. 2006, 23, 2611–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdurasulova, I.N.; Serdyuk, S.E.; Gmiro, V.E. Comparative study of preventive and therapeutic effects of IEM-1966 and memantine in rats with experimental allergic encephalomyelitis. Bull. Exp. Biol. Med. 2007, 144, 217–220. [Google Scholar] [CrossRef]
- Schuh, C.; Wimmer, I.; Hametner, S.; Haider, L.; Van Dam, A.-M.; Liblau, R.S.; Smith, K.J.; Probert, L.; Binder, C.J.; Bauer, J.; et al. Oxidative tissue injury in multiple sclerosis is only partly reflected in experimental disease models. Acta Neuropathol. 2014, 128, 247–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Ljubisavljevic, S.; Stojanovic, I.; Pavlovic, D.; Sokolovic, D.; Stevanovic, I. Aminoguanidine and N-acetyl-cysteine supress oxidative and nitrosative stress in EAE rat brains. Redox Rep. 2011, 16, 166–172. [Google Scholar] [CrossRef]
- Ljubisavljevic, S.; Stojanovic, I.; Pavlovic, D.; Milojkovic, M.; Sokolovic, D.; Stevanovic, I.; Petrovic, A. Suppression of the lipid peroxidation process in the CNS reduces neurological expression of experimentally induced autoimmune encephalomyelitis. Folia Neuropathol. 2013, 51, 51–57. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-Mediated Inactivation of Manganese Superoxide Dismutase Involves Nitration and Oxidation of Critical Tyrosine Residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Kerschensteiner, M.; Stadelmann, C.; Buddeberg, B.S.; Merkler, D.; Bareyre, F.M.; Anthony, D.C.; Linington, C.; Brück, W.; Schwab, M.E. Targeting Experimental Autoimmune Encephalomyelitis Lesions to a Predetermined Axonal Tract System Allows for Refined Behavioral Testing in an Animal Model of Multiple Sclerosis. Am. J. Pathol. 2004, 164, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.; Weissert, R.; Diem, R.; Storch, M.K.; de Graaf, K.L.; Kramer, B.; Bähr, M. Acute Neuronal Apoptosis in a Rat Model of Multiple Sclerosis. J. Neurosci. 2001, 21, 6214–6220. [Google Scholar] [CrossRef]
- Ohgoh, M.; Hanada, T.; Smith, T.; Hashimoto, T.; Ueno, M.; Yamanishi, Y.; Watanabe, M.; Nishizawa, Y. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 125, 170–178. [Google Scholar] [CrossRef]
- Asakawa, T.; Matsushita, S. Coloring conditions of thiobarbituric acid test for detecting lipid hydroperoxides. Lipids 1980, 15, 137–140. [Google Scholar] [CrossRef]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosenbrough, N.J.; Farr, A.L.; Randal, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Body Weight (g) | |||
|---|---|---|---|
| Days Post Immunization (d.p.i.) | Control | EAE | EAE + Memantine |
| 0 | 194.0 ± 8.7 | 193.6 ± 10.4 | 195.9 ± 11.8 |
| 4 | 198.4 ± 10.8 | 190.4 ± 10.1 | 191.6 ± 9.1 |
| 12 | 210.7 ± 4.3 | 157.9 ± 9.9 *** | 166.0 ± 10.3 *** |
| 20 | 212.0 ± 3.3 | 163.2 ± 7.7 *** | 182.1 ± 5.1 ***,### |
| 25 | 211.8 ± 2.6 | 172.0 ± 12.9 *** | 188.4 ± 8.8 **,# |
| Characteristics/Clinical Parameters | EAE | EAE + Memantine |
|---|---|---|
| Animals with clinical symptoms (%) | 100 | 98.44 |
| Animals with severe EAE (%) | 76.67 | 61.11 |
| Lethality (%) | 3.33 | 2.22 |
| Inductive phase (days) | 9.5 ± 1.4 | 11.3 ± 0.8 *** |
| Cumulative CI (score) | 16.93 ± 1.16 | 9.57 ± 0.67 *** |
| Duration of disease (days) | 17.5 ± 1.4 | 15.2 ± 1.0 *** |
| Number of animals | 90 | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dąbrowska-Bouta, B.; Strużyńska, L.; Sidoryk-Węgrzynowicz, M.; Sulkowski, G. Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2021, 22, 11330. https://doi.org/10.3390/ijms222111330
Dąbrowska-Bouta B, Strużyńska L, Sidoryk-Węgrzynowicz M, Sulkowski G. Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2021; 22(21):11330. https://doi.org/10.3390/ijms222111330
Chicago/Turabian StyleDąbrowska-Bouta, Beata, Lidia Strużyńska, Marta Sidoryk-Węgrzynowicz, and Grzegorz Sulkowski. 2021. "Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 22, no. 21: 11330. https://doi.org/10.3390/ijms222111330
APA StyleDąbrowska-Bouta, B., Strużyńska, L., Sidoryk-Węgrzynowicz, M., & Sulkowski, G. (2021). Memantine Modulates Oxidative Stress in the Rat Brain following Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 22(21), 11330. https://doi.org/10.3390/ijms222111330