
The Structure of the Electric Double Layer of the Protic Ionic Liquid [Dema][TfO] Analyzed by Atomic Force Spectroscopy

 ,
,  , ,
, , 

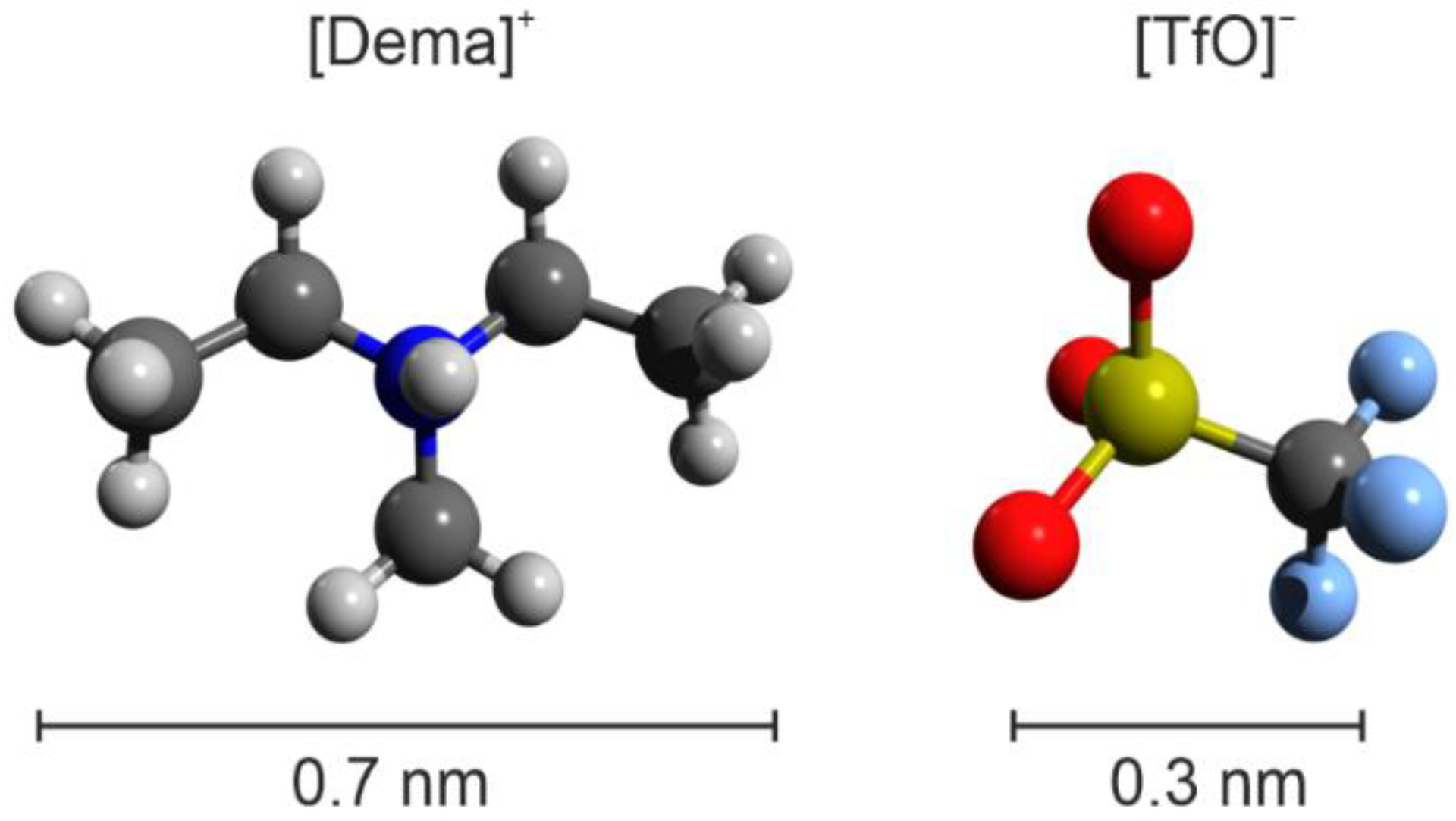{kind=link}
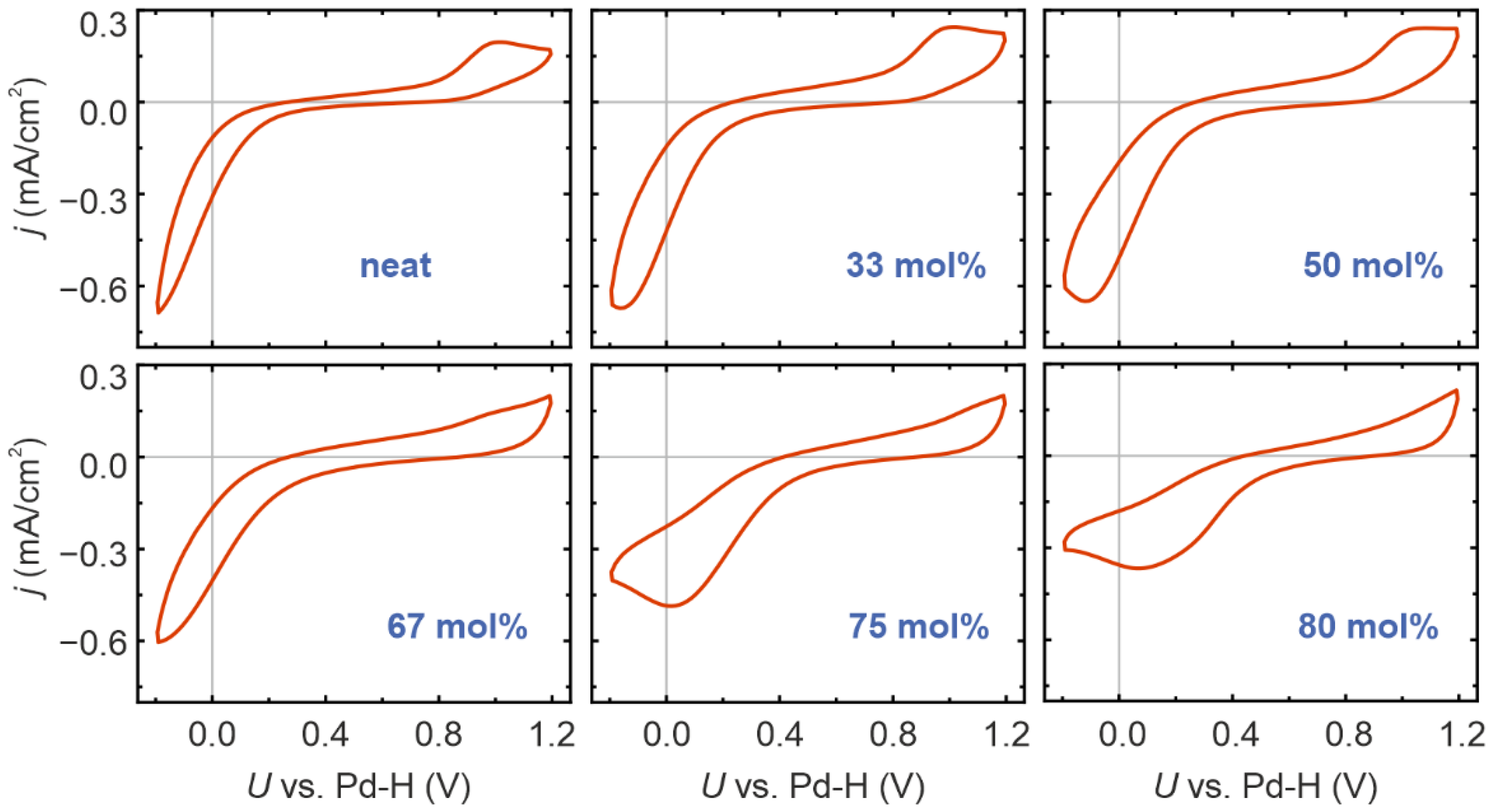{kind=link}
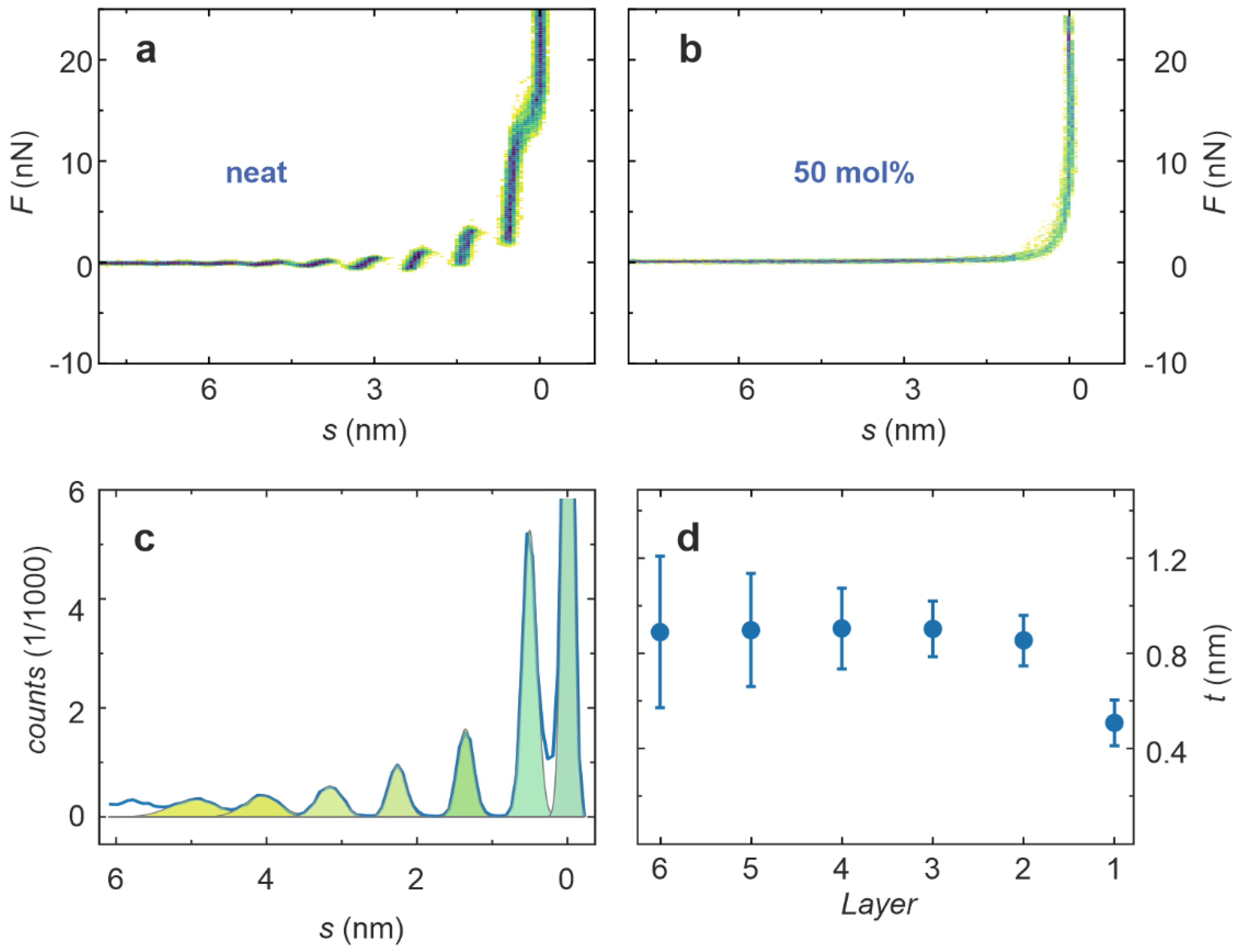{kind=link}
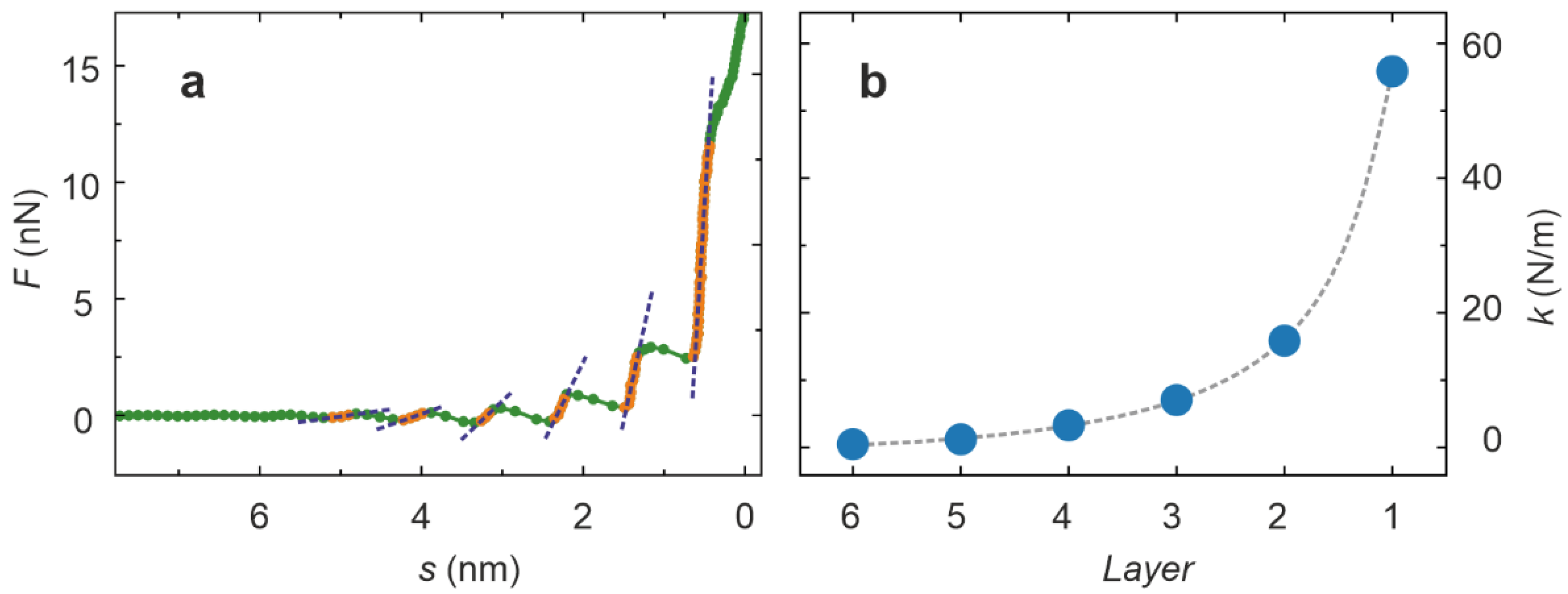{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
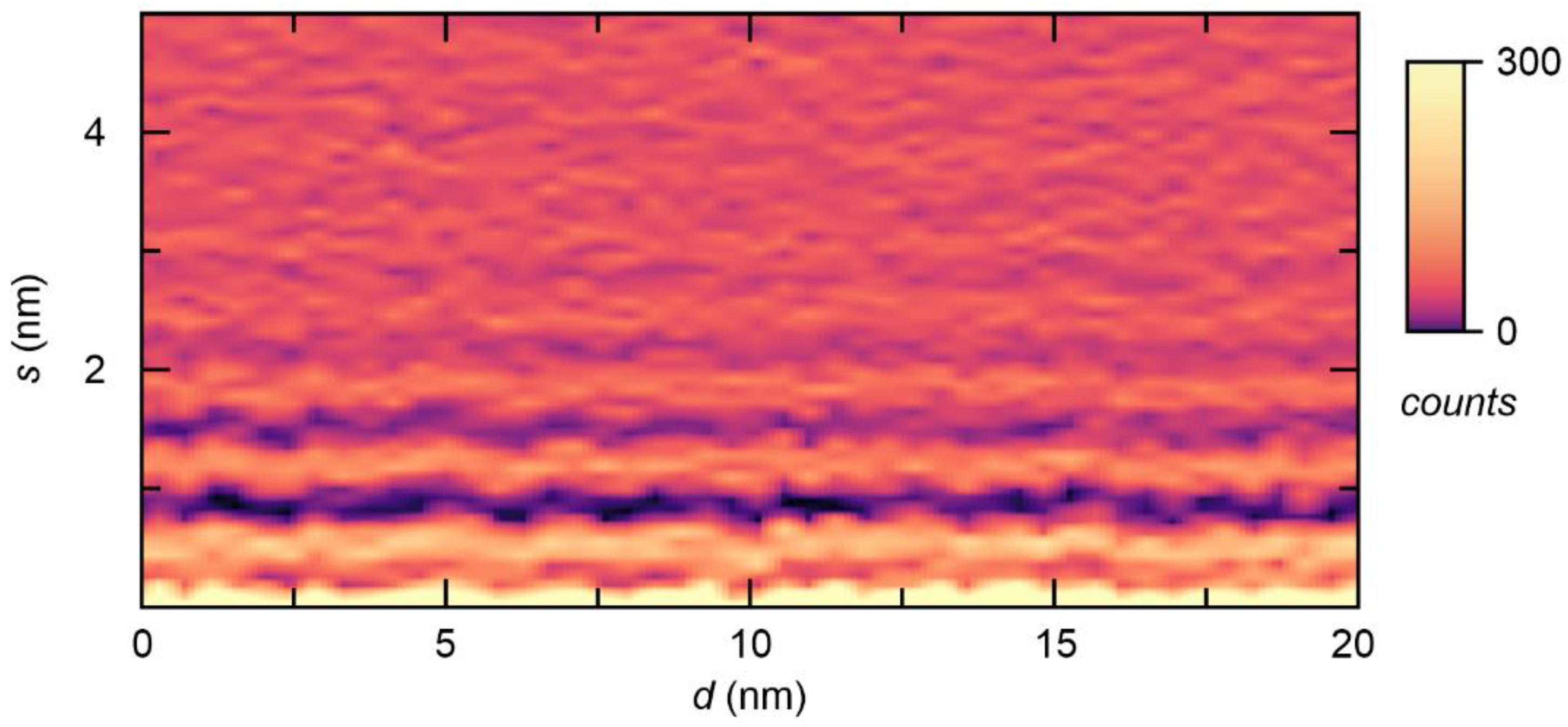
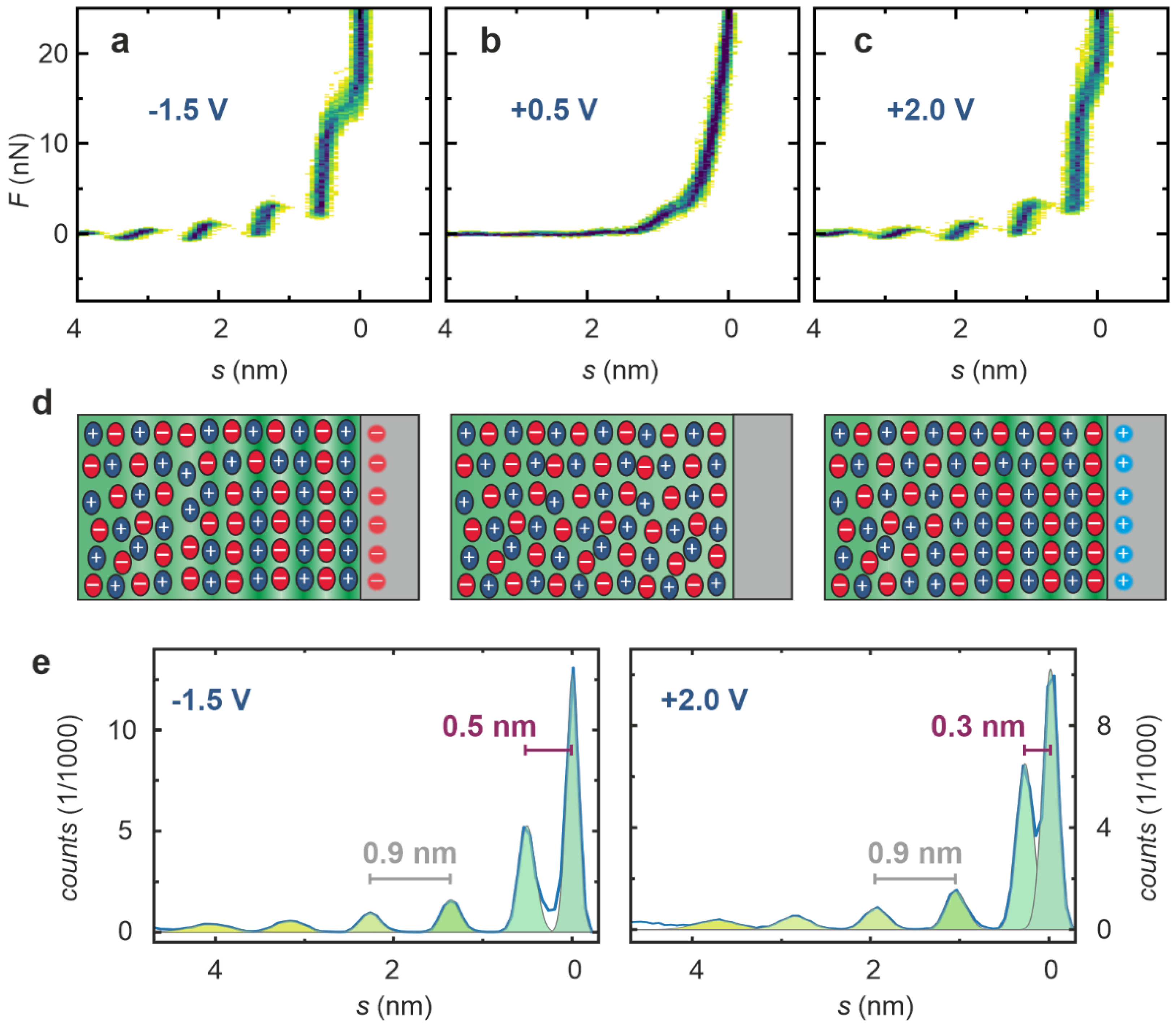
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yasuda, T.; Watanabe, M. Protic Ionic Liquids: Fuel Cell Applications. MRS Bull. 2013, 38, 560–566. [Google Scholar] [CrossRef]
- Wippermann, K.; Wackerl, J.; Lehnert, W.; Huber, B.; Korte, C. 2-Sulfoethylammonium Trifluoromethanesulfonate as an Ionic Liquid for High Temperature PEM Fuel Cells. J. Electrochem. Soc. 2016, 163, F25–F37. [Google Scholar] [CrossRef] [Green Version]
- Branco, C.M.; Sharma, S.; de Camargo Forte, M.M.; Steinberger-Wilckens, R. New Approaches towards Novel Composite and Multilayer Membranes for Intermediate Temperature-Polymer Electrolyte Fuel Cells and Direct Methanol Fuel Cells. J. Power Sources 2016, 316, 139–159. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, M.; Kujawski, W.; Fatyeyeva, K.; Kujawa, J. A Review on Ionic Liquids-Based Membranes for Middle and High Temperature Polymer Electrolyte Membrane Fuel Cells (PEM FCs). Int. J. Mol. Sci. 2021, 22, 5430. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy Applications of Ionic Liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Ogawa, A.; Kanno, M.; Nakamoto, H.; Yasuda, T.; Watanabe, M. Nonhumidified Intermediate Temperature Fuel Cells Using Protic Ionic Liquids. J. Am. Chem. Soc. 2010, 132, 9764–9773. [Google Scholar] [CrossRef]
- Elwan, H.A.; Mamlouk, M.; Scott, K. A Review of Proton Exchange Membranes Based on Protic Ionic Liquid/Polymer Blends for Polymer Electrolyte Membrane Fuel Cells. J. Power Sources 2021, 484, 229197. [Google Scholar] [CrossRef]
- Philippi, F.; Welton, T. Targeted Modifications in Ionic Liquids from Understanding to Design. Phys. Chem. Chem. Phys. 2021, 23, 6993–7021. [Google Scholar] [CrossRef]
- Silva, W.; Zanatta, M.; Ferreira, A.S.; Corvo, M.C.; Cabrita, E.J. Revisiting Ionic Liquid Structure-Property Relationship: A Critical Analysis. Int. J. Mol. Sci. 2020, 21, 7745. [Google Scholar] [CrossRef]
- Sweeney, J.; Hausen, F.; Hayes, R.; Webber, G.B.; Endres, F.; Rutland, M.W.; Bennewitz, R.; Atkin, R. Control of Nanoscale Friction on Gold in an Ionic Liquid by a Potential-Dependent Ionic Lubricant Layer. Phys. Rev. Lett. 2012, 109, 155502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkkötter, M.; Giffin, G.A.; Moretti, A.; Jeong, S.; Passerini, S.; Schönhoff, M. Relevance of Ion Clusters for Li Transport at Elevated Salt Concentrations in [Pyr12O1][FTFSI] Ionic Liquid-Based Electrolytes. Chem. Commun. 2018, 54, 4278–4281. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.; Mailoa, J.P.; Craig, N.; Christensen, J.; Kozinsky, B. Transport Anomalies Emerging from Strong Correlation in Ionic Liquid Electrolytes. J. Power Sources 2019, 428, 27–36. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoth, J.; Hausen, F.; Müser, M.H.; Bennewitz, R. Force Microscopy of Layering and Friction in an Ionic Liquid. J. Phys. Condens. Matter 2014, 26, 284110. [Google Scholar] [CrossRef] [PubMed]
- Rodenbücher, C.; Wippermann, K.; Korte, C. Atomic Force Spectroscopy on Ionic Liquids. Appl. Sci. 2019, 9, 2207. [Google Scholar] [CrossRef] [Green Version]
- Kornyshev, A.A. Double-Layer in Ionic Liquids: Paradigm Change? J. Phys. Chem. B 2007, 111, 5545–5557. [Google Scholar] [CrossRef]
- Perkin, S. Ionic Liquids in Confined Geometries. Phys. Chem. Chem. Phys. 2012, 14, 5052. [Google Scholar] [CrossRef] [PubMed]
- Perkin, S.; Albrecht, T.; Klein, J. Layering and Shear Properties of an Ionic Liquid, 1-Ethyl-3-Methylimidazolium Ethylsulfate, Confined to Nano-Films between Mica Surfaces. Phys. Chem. Chem. Phys. 2010, 12, 1243–1247. [Google Scholar] [CrossRef]
- Mezger, M.; Schröder, H.; Reichert, H.; Schramm, S.; Okasinski, J.S.; Schöder, S.; Honkimäki, V.; Deutsch, M.; Ocko, B.M.; Ralston, J.; et al. Molecular Layering of Fluorinated Ionic Liquids at a Charged Sapphire (0001) Surface. Science 2008, 322, 424–428. [Google Scholar] [CrossRef]
- Fedorov, M.V.; Kornyshev, A.A. Ionic Liquids at Electrified Interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkin, S.; Salanne, M.; Madden, P.; Lynden-Bell, R. Is a Stern and Diffuse Layer Model Appropriate to Ionic Liquids at Surfaces? Proc. Natl. Acad. Sci. USA 2013, 110, E4121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.M.; Zhu, M.; Zhang, P.; Unocic, R.R.; Guo, D.; Okatan, M.B.; Dai, S.; Cummings, P.T.; Kalinin, S.V.; Feng, G.; et al. Fundamental Aspects of Electric Double Layer Force-Distance Measurements at Liquid-Solid Interfaces Using Atomic Force Microscopy. Sci. Rep. 2016, 6, 32389. [Google Scholar] [CrossRef] [PubMed]
- Jurado, L.A.; Kim, H.; Rossi, A.; Arcifa, A.; Schuh, J.K.; Spencer, N.D.; Leal, C.; Ewoldt, R.H.; Espinosa-Marzal, R.M. Effect of the Environmental Humidity on the Bulk, Interfacial and Nanoconfined Properties of an Ionic Liquid. Phys. Chem. Chem. Phys. 2016, 18, 22719–22730. [Google Scholar] [CrossRef]
- Espinosa-Marzal, R.M.; Arcifa, A.; Rossi, A.; Spencer, N.D. Microslips to “Avalanches” in Confined, Molecular Layers of Ionic Liquids. J. Phys. Chem. Lett. 2014, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, Y.; Sun, X.; Zhang, Z.; Mu, T. Water Sorption in Ionic Liquids: Kinetics, Mechanisms and Hydrophilicity. Phys. Chem. Chem. Phys. 2012, 14, 12252. [Google Scholar] [CrossRef] [PubMed]
- Fatyeyeva, K.; Rogalsky, S.; Makhno, S.; Tarasyuk, O.; Soto Puente, J.A.; Marais, S. Polyimide/Ionic Liquid Composite Membranes for Middle and High Temperature Fuel Cell Application: Water Sorption Behavior and Proton Conductivity. Membranes 2020, 10, 82. [Google Scholar] [CrossRef]
- Bi, S.; Wang, R.; Liu, S.; Yan, J.; Mao, B.; Kornyshev, A.A.; Feng, G. Minimizing the Electrosorption of Water from Humid Ionic Liquids on Electrodes. Nat. Commun. 2018, 9, 5222. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Yan, J.; Li, M.; Chen, L.; Mao, B. The Electric Double Layer in an Ionic Liquid Incorporated with Water Molecules: Atomic Force Microscopy Force Curve Study. ChemElectroChem 2016, 3, 2221–2226. [Google Scholar] [CrossRef]
- Kemna, A.; Braunschweig, B. Potential-Induced Adsorption and Structuring of Water at the Pt(111) Electrode Surface in Contact with an Ionic Liquid. J. Phys. Chem. Lett. 2020, 11, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-W.; Dienemann, J.-N.; Stock, P.; Merola, C.; Chen, Y.-J.; Valtiner, M. The Effect of Water and Confinement on Self-Assembly of Imidazolium Based Ionic Liquids at Mica Interfaces. Sci. Rep. 2016, 6, 30058. [Google Scholar] [CrossRef] [PubMed]
- Thimmappa, R.; Walsh, D.; Scott, K.; Mamlouk, M. Diethylmethylammonium Trifluoromethanesulfonate Protic Ionic Liquid Electrolytes for Water Electrolysis. J. Power Sources 2020, 449, 227602. [Google Scholar] [CrossRef]
- Smith, D.E.; Walsh, D.A. The Nature of Proton Shuttling in Protic Ionic Liquid Fuel Cells. Adv. Energy Mater. 2019, 9, 1900744. [Google Scholar] [CrossRef]
- Lin, J.; Wang, L.; Zinkevich, T.; Indris, S.; Suo, Y.; Korte, C. Influence of Residual Water and Cation Acidity on the Ionic Transport Mechanism in Proton-Conducting Ionic Liquids. Phys. Chem. Chem. Phys. 2020, 22, 1145–1153. [Google Scholar] [CrossRef]
- Wippermann, K.; Suo, Y.; Korte, C. Oxygen Reduction Reaction Kinetics on Pt in Mixtures of Proton-Conducting Ionic Liquids and Water: The Influence of Cation Acidity. J. Phys. Chem. C 2021, 125, 4423–4435. [Google Scholar] [CrossRef]
- Wippermann, K.; Giffin, J.; Korte, C. In Situ Determination of the Water Content of Ionic Liquids. J. Electrochem. Soc. 2018, 165, H263–H270. [Google Scholar] [CrossRef]
- Wippermann, K.; Giffin, J.; Kuhri, S.; Lehnert, W.; Korte, C. The Influence of Water Content in a Proton-Conducting Ionic Liquid on the Double Layer Properties of the Pt/PIL Interface. Phys. Chem. Chem. Phys. 2017, 19, 24706–24723. [Google Scholar] [CrossRef]
- Walsh, D.A.; Ejigu, A.; Smith, J.; Licence, P. Kinetics and Mechanism of Oxygen Reduction in a Protic Ionic Liquid. Phys. Chem. Chem. Phys. 2013, 15, 7548. [Google Scholar] [CrossRef] [PubMed]
- Black, J.M.; Baris Okatan, M.; Feng, G.; Cummings, P.T.; Kalinin, S.V.; Balke, N. Topological Defects in Electric Double Layers of Ionic Liquids at Carbon Interfaces. Nano Energy 2015, 15, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Suo, Y.; Hou, H.; Lin, J.; Chen, Y.; Liu, C.; Wang, C.; Schulz, P.S.; Wippermann, K.; Korte, C. Binary Mixtures of Proton-Conducting Ionic Liquids as Electrolytes for Medium-Temperature Polymer Electrolyte Membrane Fuel Cells. J. Phys. Chem. C 2021, 125, 39. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- LAMMPS Molecular Dynamics Simulator. Available online: www.lammps.org (accessed on 28 October 2021).
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Nasrabadi, A.T.; Gelb, L.D. Structural and Transport Properties of Tertiary Ammonium Triflate Ionic Liquids: A Molecular Dynamics Study. J. Phys. Chem. B 2017, 121, 1908–1921. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible Simple Point-Charge Water Model with Improved Liquid-State Properties. J. Chem. Phys. 2006, 124, 024503. [Google Scholar] [CrossRef]
- Liem, S.Y.; Chan, K.-Y. Effective Pairwise Potential for Simulations of Adsorbed Platinum. Mol. Phys. 1995, 86, 939–949. [Google Scholar] [CrossRef]
- Merkel, N.C.; Römich, C.; Bernewitz, R.; Künemund, H.; Gleiß, M.; Sauer, S.; Schubert, T.J.S.; Guthausen, G.; Schaber, K. Thermophysical Properties of the Binary Mixture of Water + Diethylmethylammonium Trifluoromethanesulfonate and the Ternary Mixture of Water + Diethylmethylammonium Trifluoromethanesulfonate + Diethylmethylammonium Methanesulfonate. J. Chem. Eng. Data 2014, 59, 560–570. [Google Scholar] [CrossRef]
- Yang, J.; Bo, Z.; Yang, H.; Qi, H.; Kong, J.; Yan, J.; Cen, K. Reliability of Constant Charge Method for Molecular Dynamics Simulations on EDLCs in Nanometer and Sub-Nanometer Spaces. ChemElectroChem 2017, 4, 2486–2493. [Google Scholar] [CrossRef]
- Noh, C.; Jung, Y. Understanding the Charging Dynamics of an Ionic Liquid Electric Double Layer Capacitor via Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2019, 21, 6790–6800. [Google Scholar] [CrossRef] [PubMed]
- Wiebe, J.; Spohr, E. Double Layer Effects in a Model of Proton Discharge on Charged Electrodes. Beilstein J. Nanotechnol. 2014, 5, 973–982. [Google Scholar] [CrossRef]
- Tesch, R.; Kowalski, P.M.; Eikerling, M.H. Properties of the Pt(111)/Electrolyte Electrochemical Interface Studied with a Hybrid DFT-Solvation Approach. J. Phys. Condens. Matter 2021, 33, 444004. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodenbücher, C.; Chen, Y.; Wippermann, K.; Kowalski, P.M.; Giesen, M.; Mayer, D.; Hausen, F.; Korte, C. The Structure of the Electric Double Layer of the Protic Ionic Liquid [Dema][TfO] Analyzed by Atomic Force Spectroscopy. Int. J. Mol. Sci. 2021, 22, 12653. https://doi.org/10.3390/ijms222312653
Rodenbücher C, Chen Y, Wippermann K, Kowalski PM, Giesen M, Mayer D, Hausen F, Korte C. The Structure of the Electric Double Layer of the Protic Ionic Liquid [Dema][TfO] Analyzed by Atomic Force Spectroscopy. International Journal of Molecular Sciences. 2021; 22(23):12653. https://doi.org/10.3390/ijms222312653
Chicago/Turabian StyleRodenbücher, Christian, Yingzhen Chen, Klaus Wippermann, Piotr M. Kowalski, Margret Giesen, Dirk Mayer, Florian Hausen, and Carsten Korte. 2021. "The Structure of the Electric Double Layer of the Protic Ionic Liquid [Dema][TfO] Analyzed by Atomic Force Spectroscopy" International Journal of Molecular Sciences 22, no. 23: 12653. https://doi.org/10.3390/ijms222312653
APA StyleRodenbücher, C., Chen, Y., Wippermann, K., Kowalski, P. M., Giesen, M., Mayer, D., Hausen, F., & Korte, C. (2021). The Structure of the Electric Double Layer of the Protic Ionic Liquid [Dema][TfO] Analyzed by Atomic Force Spectroscopy. International Journal of Molecular Sciences, 22(23), 12653. https://doi.org/10.3390/ijms222312653