
DOPA Homeostasis by Dopamine: A Control-Theoretic View



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
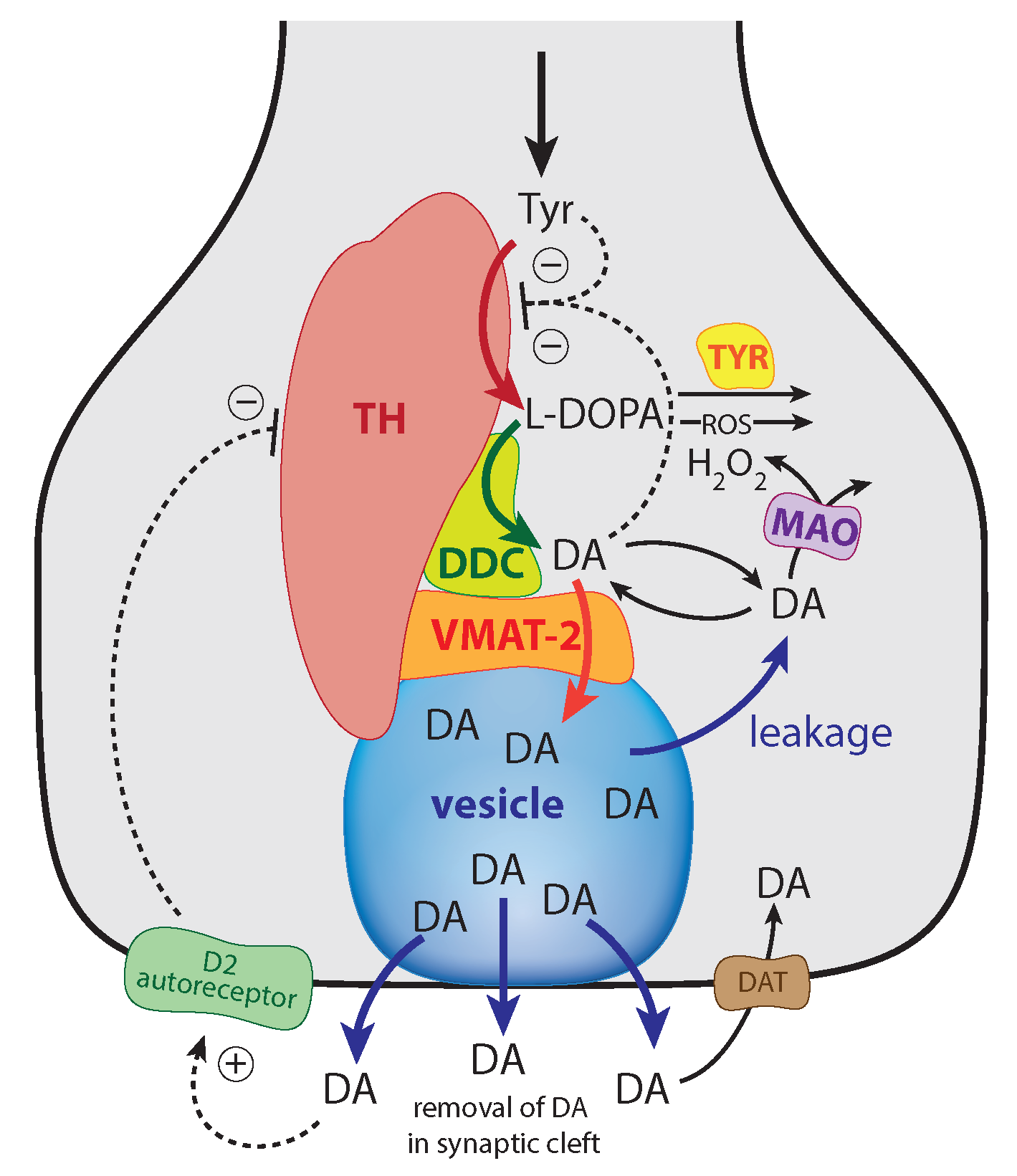
:1. Introduction
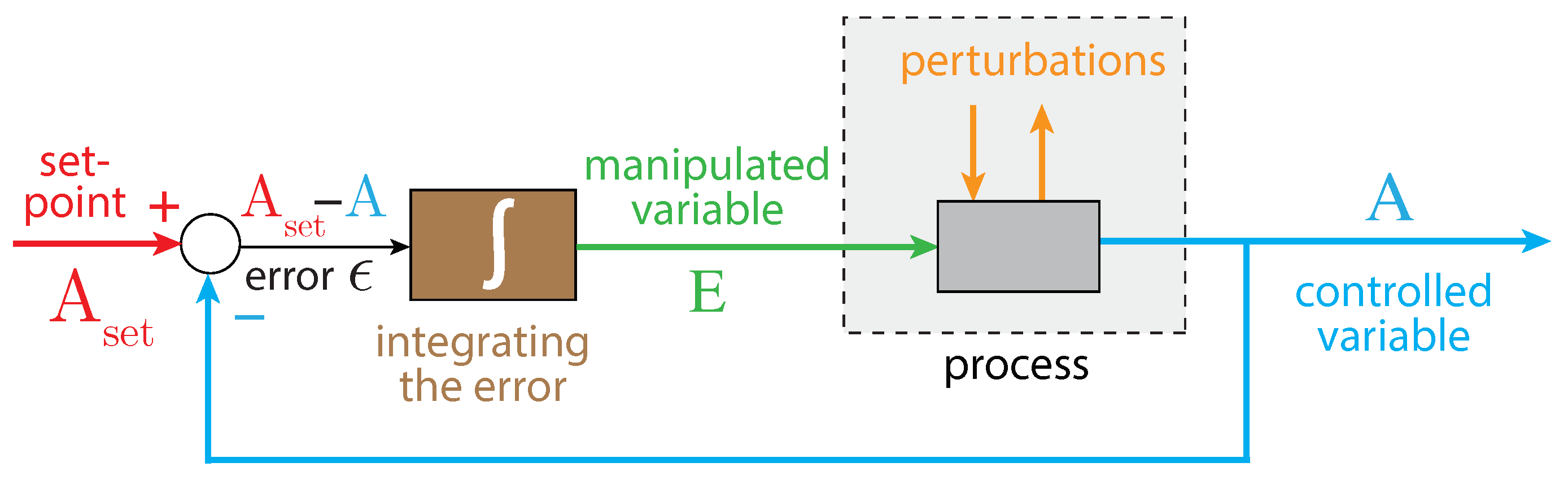
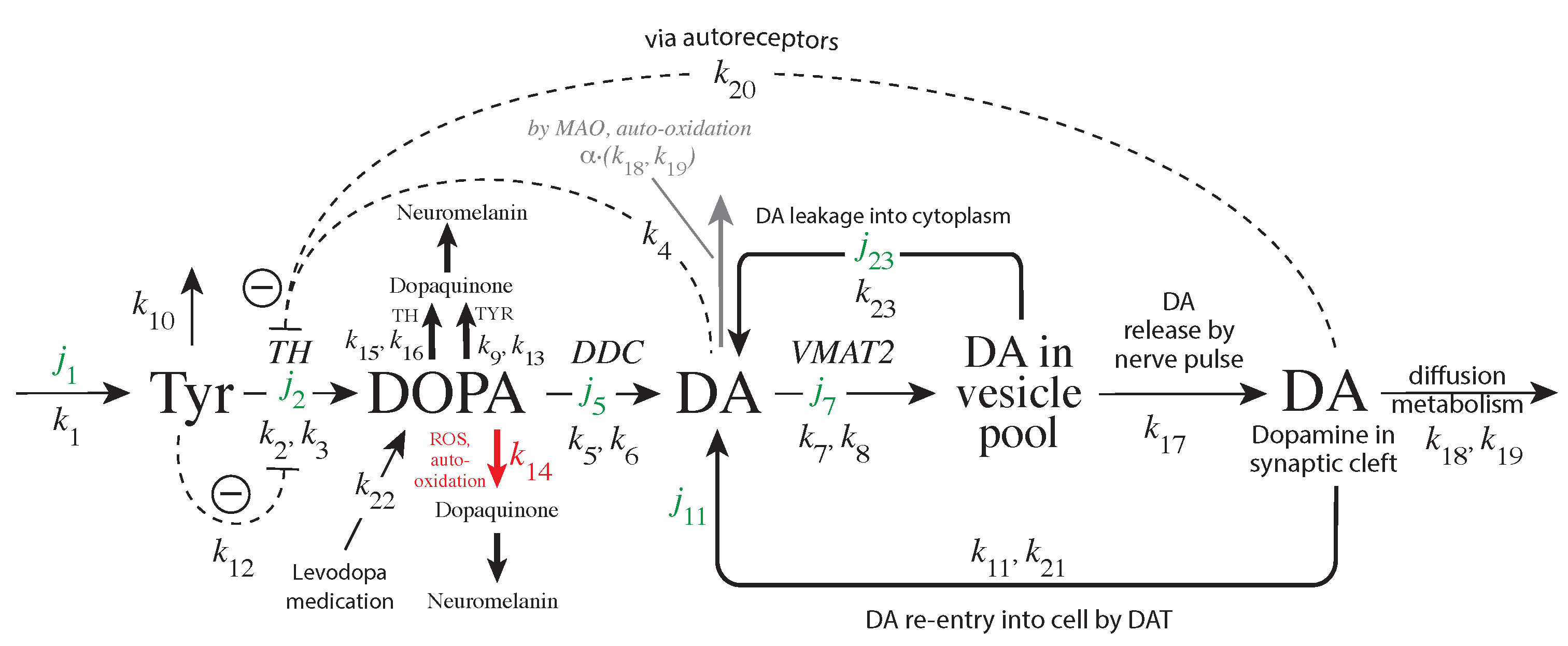
Mechanisms Leading to Robust Homeostasis
2. Materials and Methods
2.1. Method of Calculation
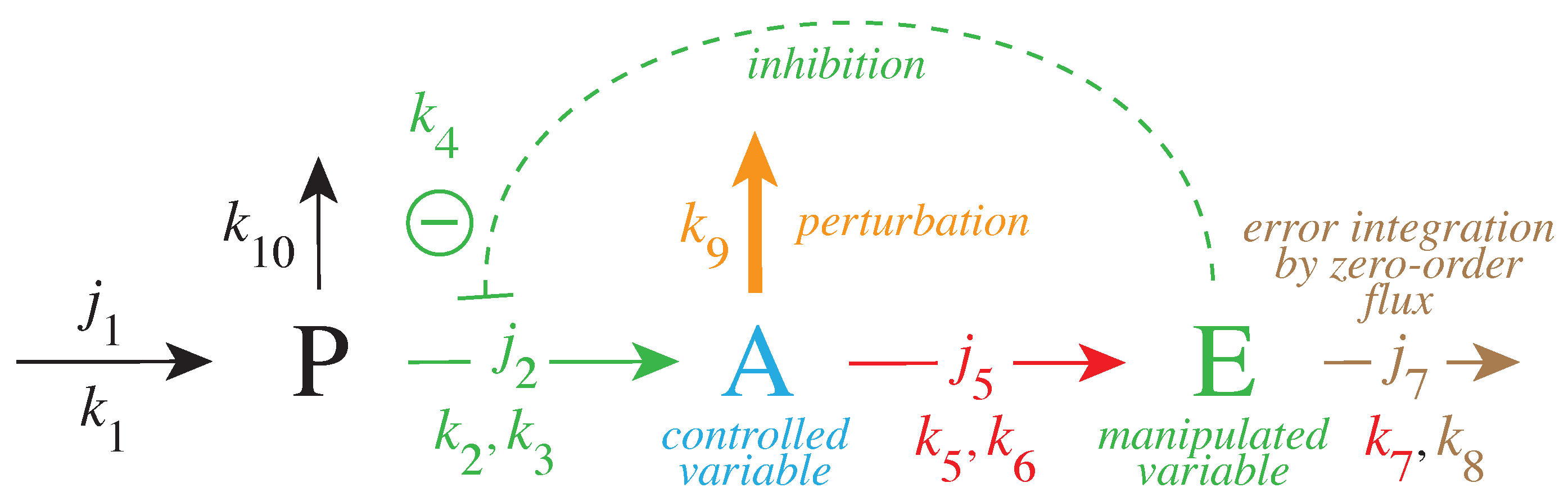
2.2. Model of Tyrosine Hydroxylase Regulation
2.3. Parameter Values
2.3.1. Dopamine Transport into Vesicles and Leakage
2.3.2. Dopamine Re-Entry by DAT
2.3.3. DOPA Decarboxylase (DCC) EC 4.1.1.28
2.3.4. Tyrosinase (TYR) EC 1.14.18.1
2.3.5. Tyrosine Hydroxylase (TH) EC 1.14.16.2. Inhibition by Tyr
2.3.6. Inhibition of TH by DA
2.3.7. Inhibition of TH by DOPA
2.3.8. TH Turnover Number/ for Tyr as Substrate
2.3.9. TH (Tyr) Values,
2.3.10. TH-Mediated Conversion of DOPA
2.3.11. Monoamine Oxidase (MAO) EC 1.4.3.4
3. Results
3.1. Factors Influencing DOPA Homeostasis
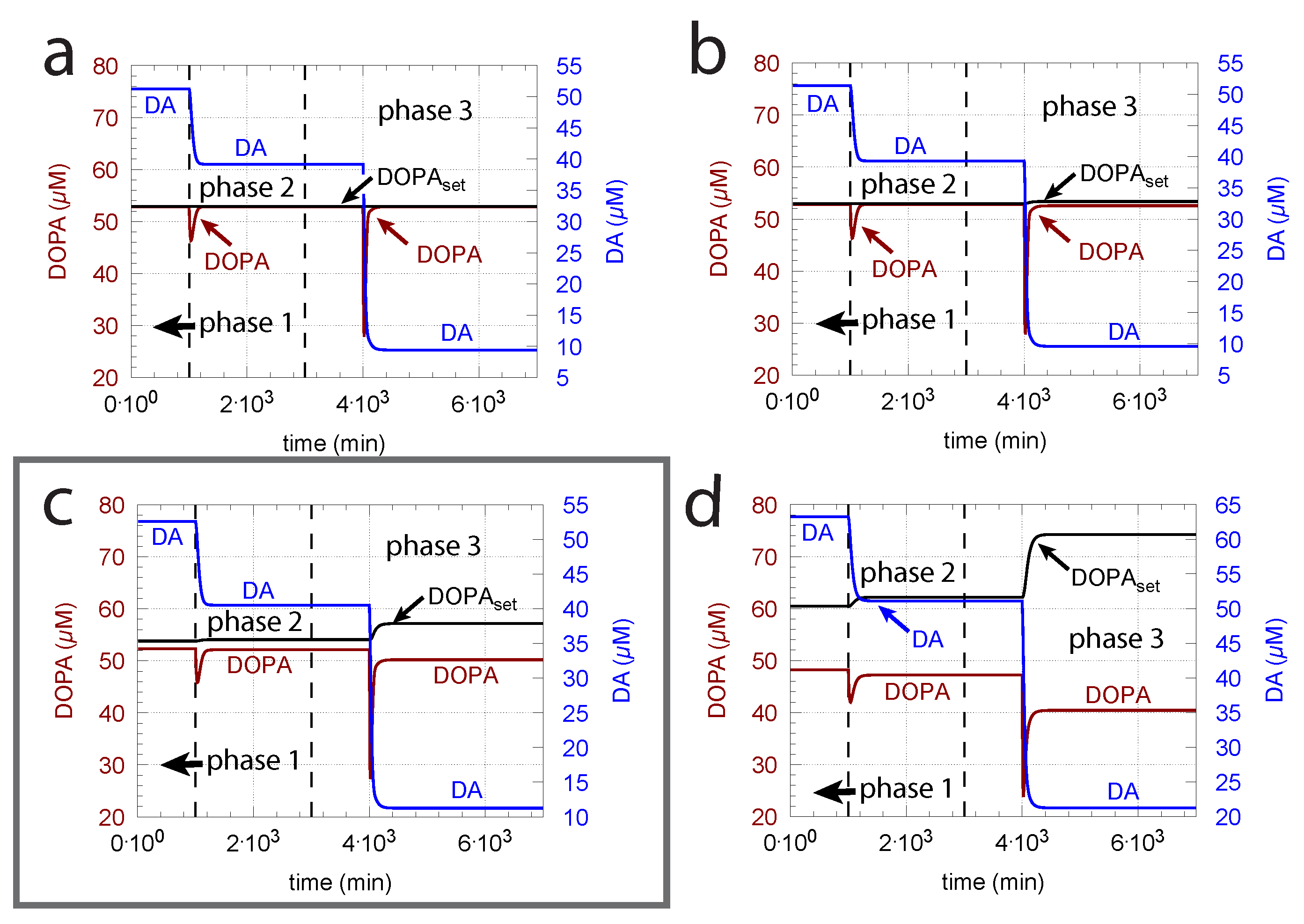
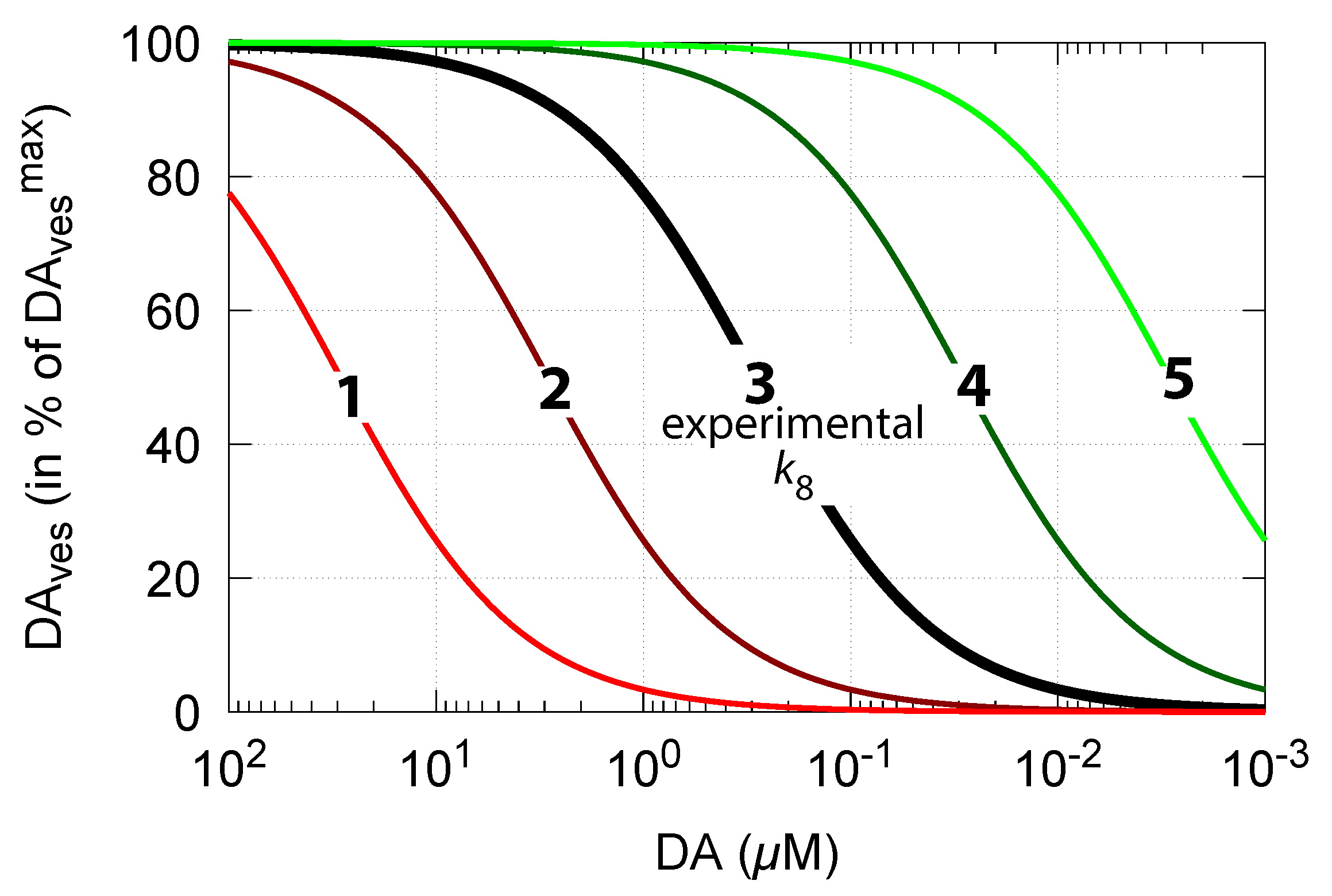
3.1.1. of Dopamine Loading into Vesicles
3.1.2. Influence of Compensatory Flux
3.1.3. TH Inhibition by DA
3.1.4. Levodopa Treatment
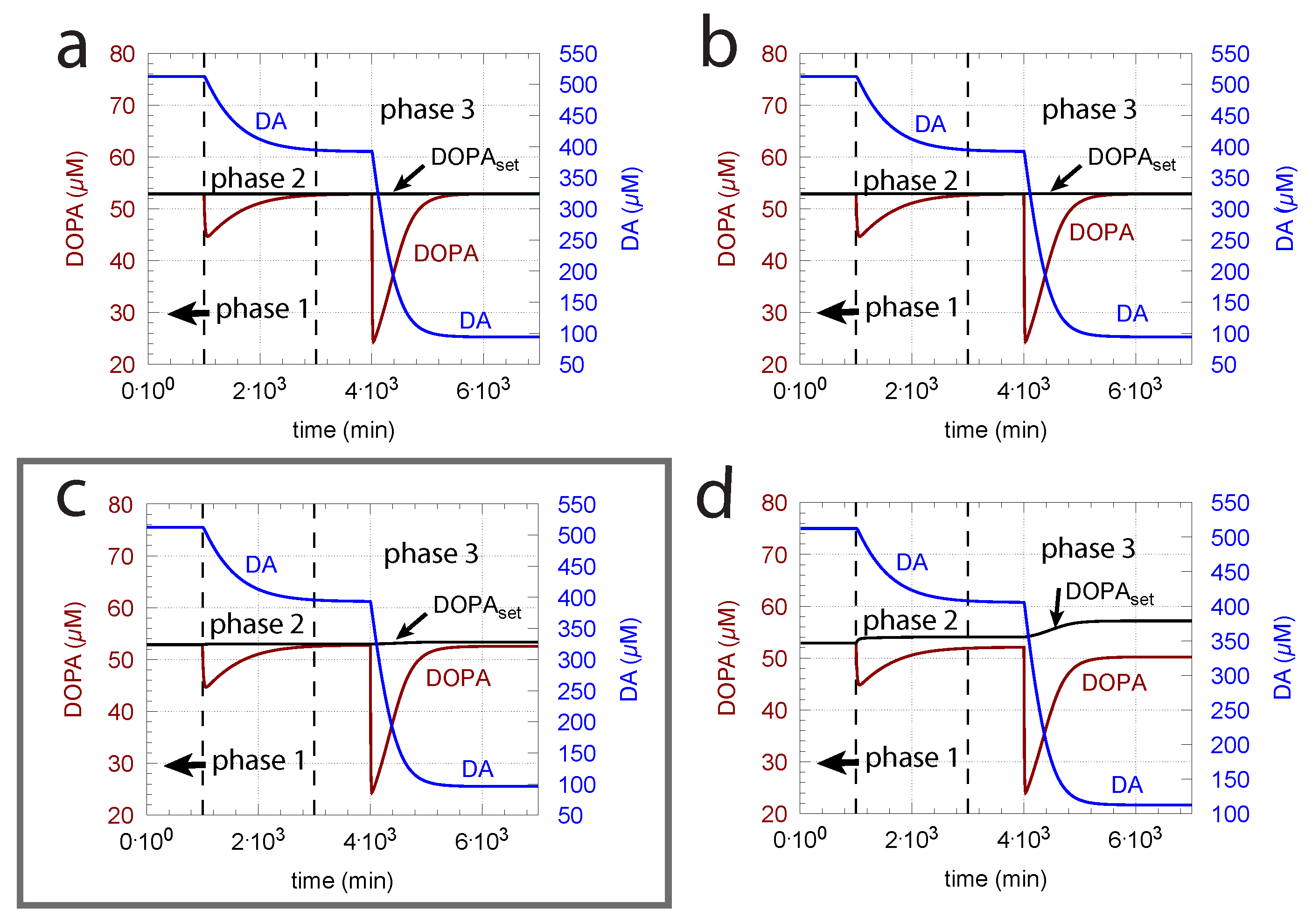
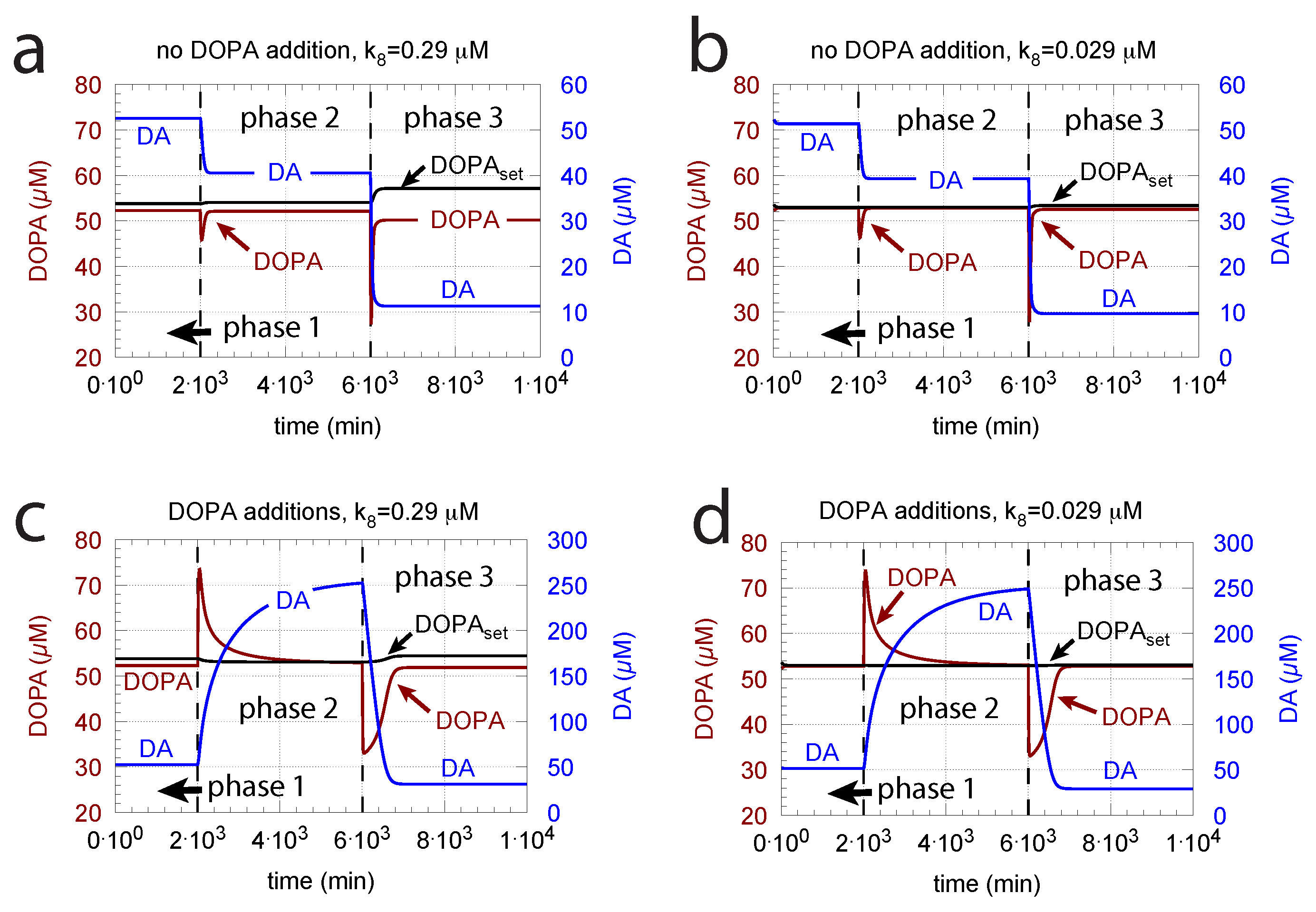
- (i)
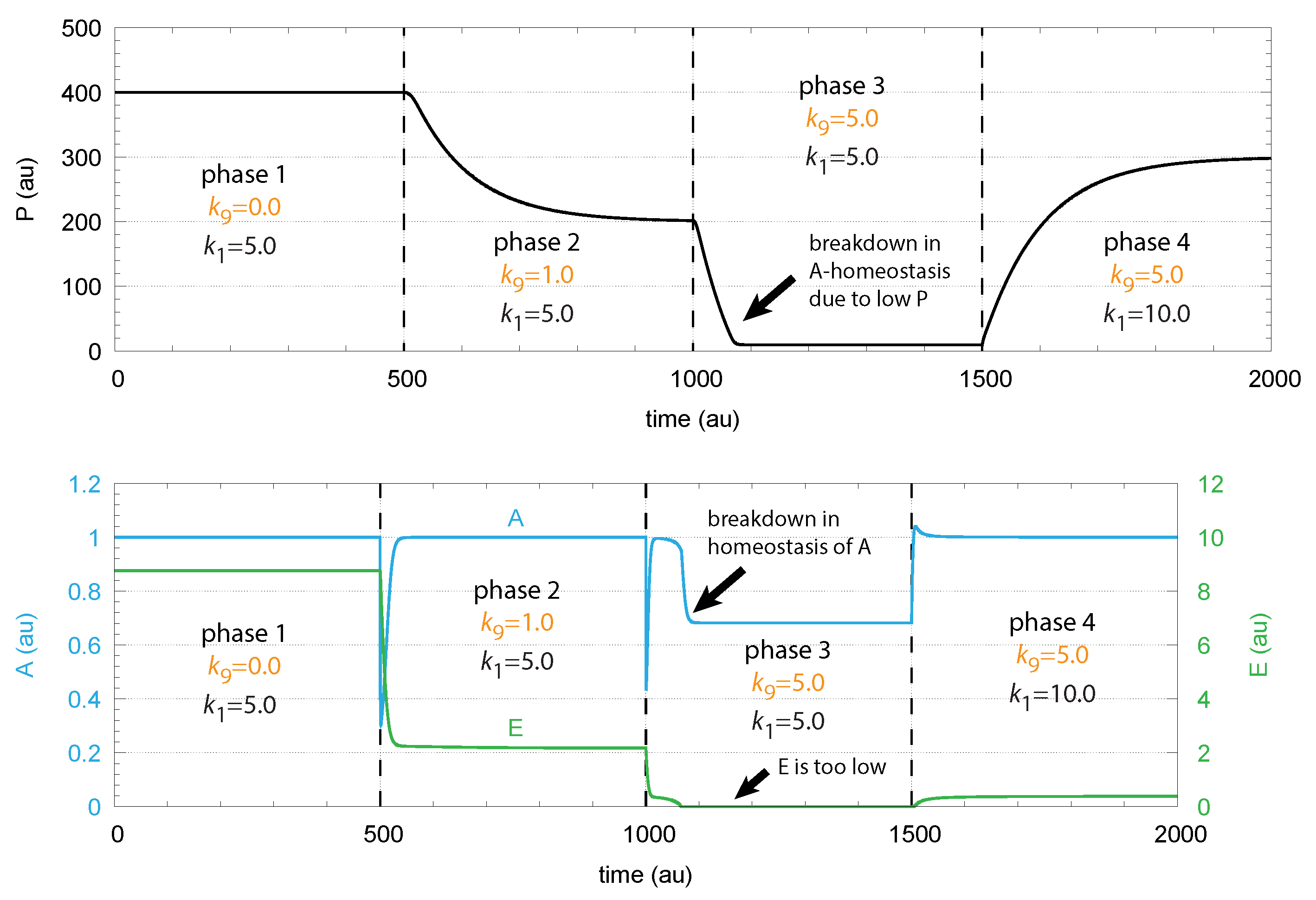
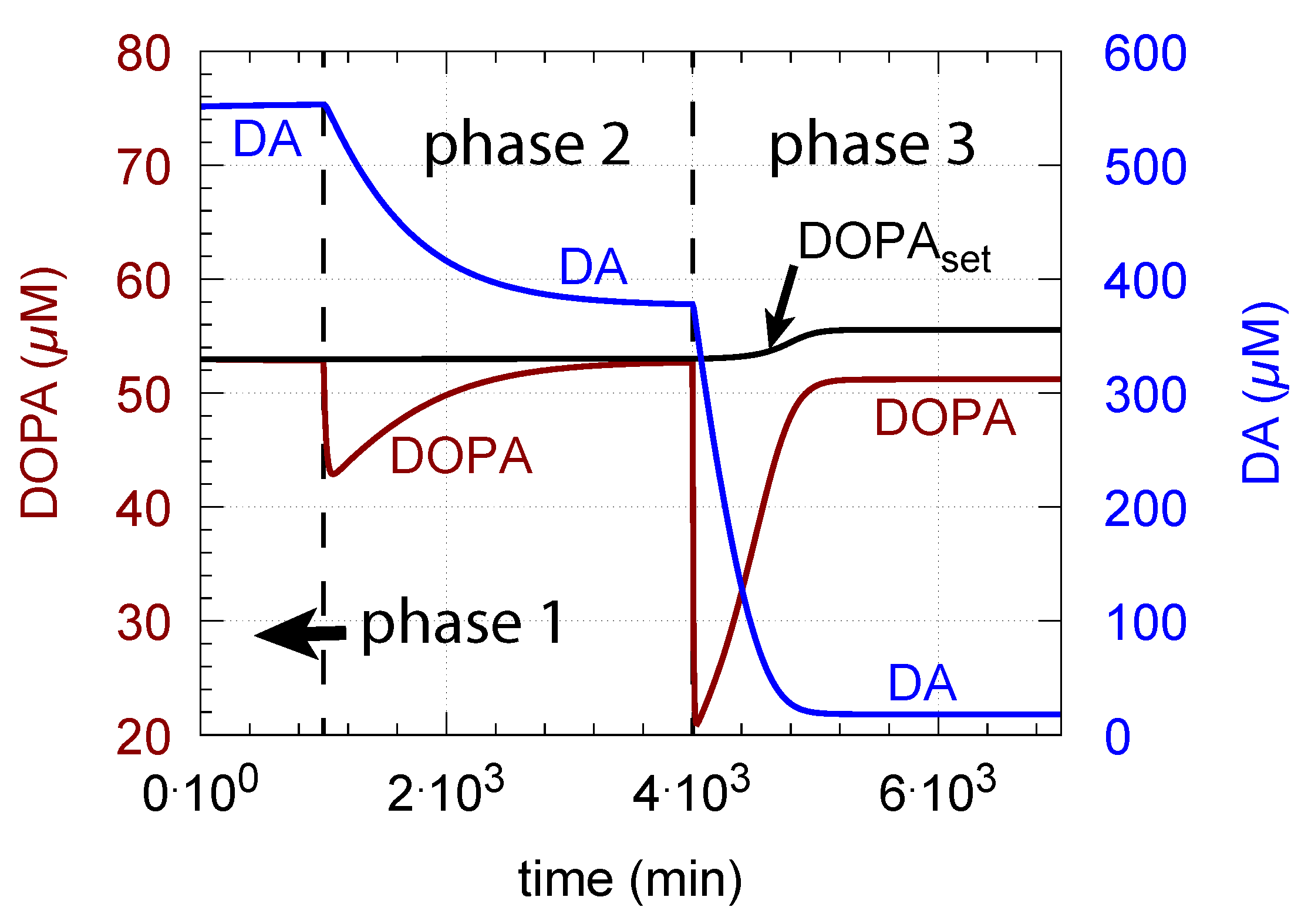
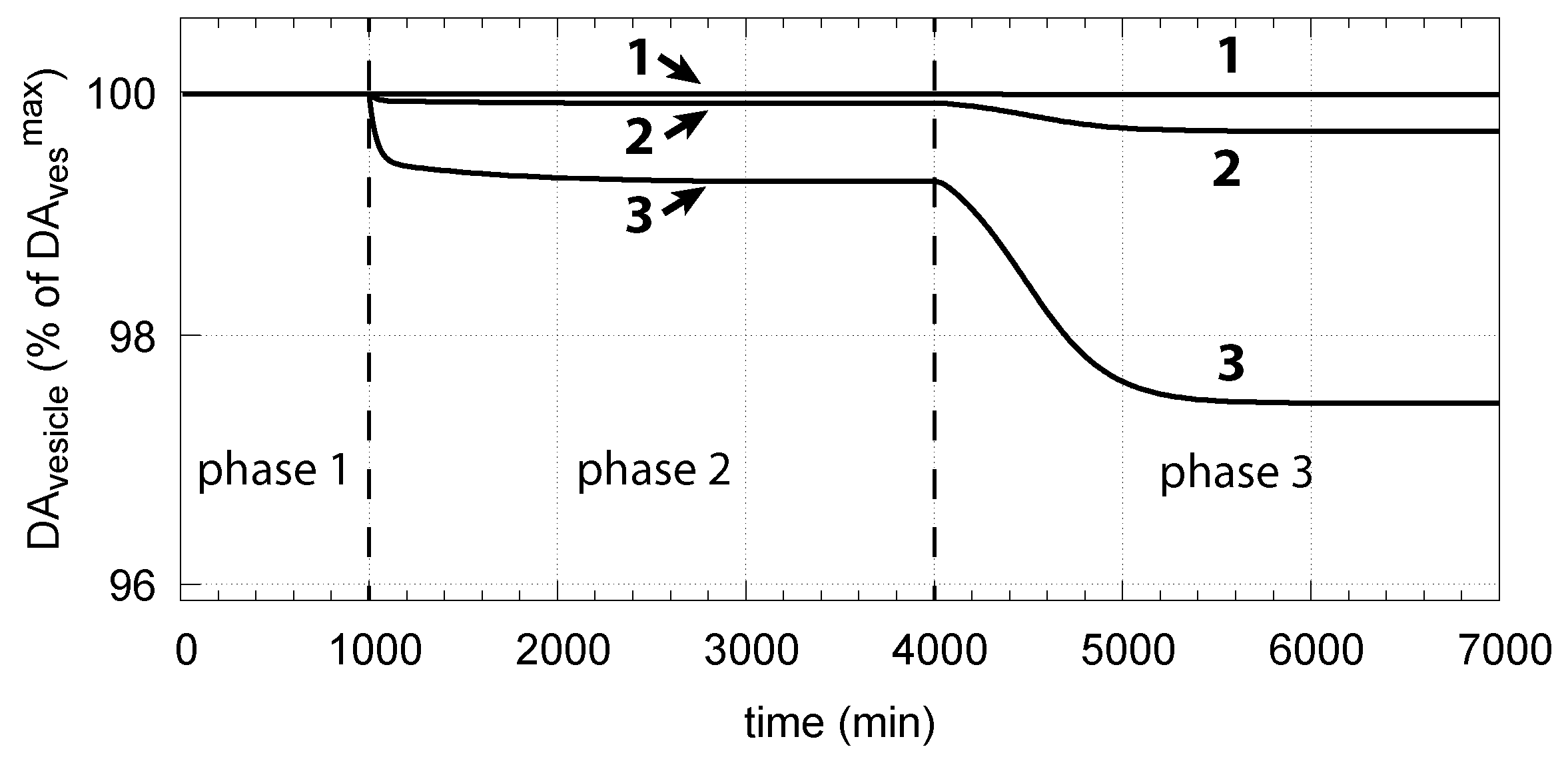
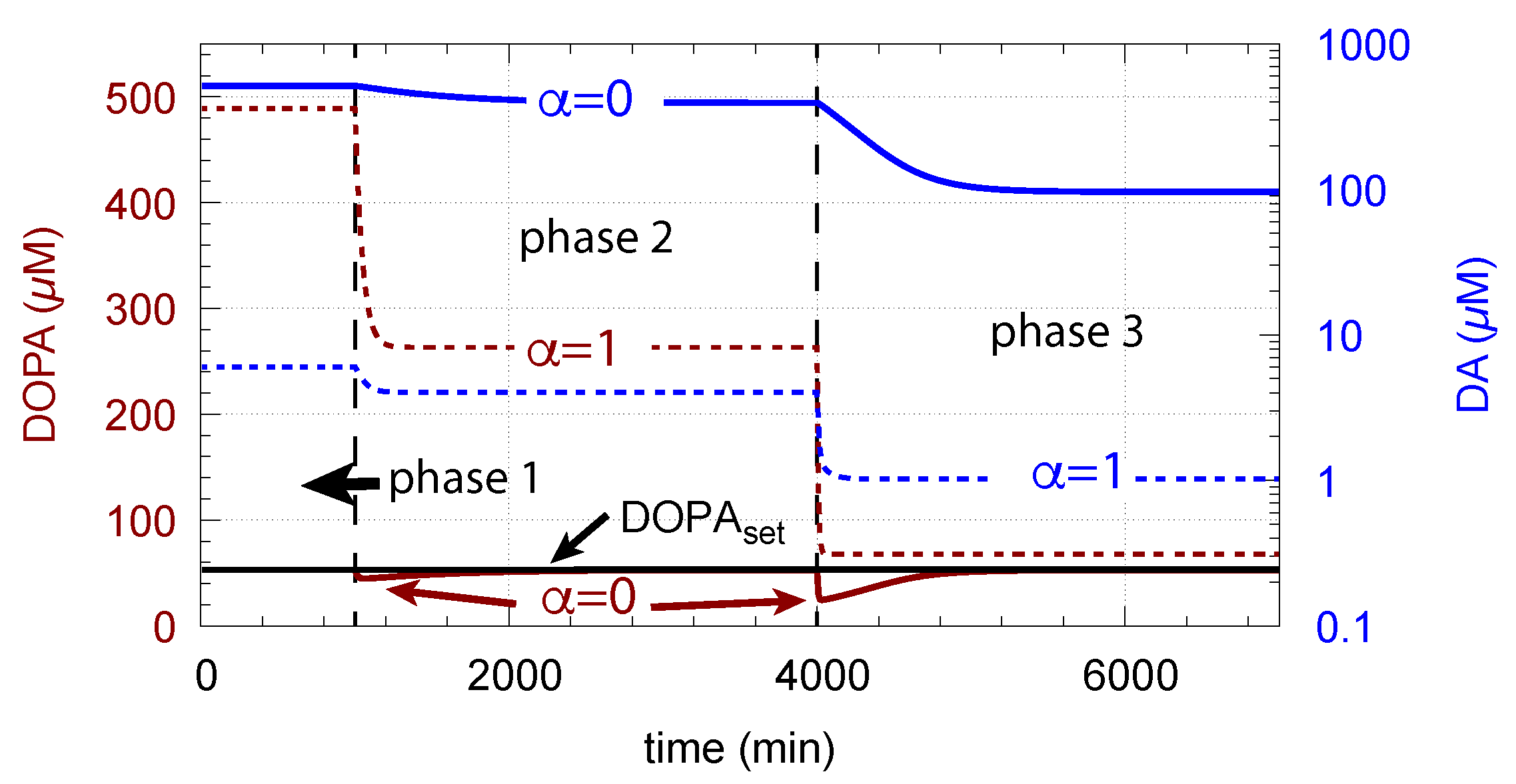
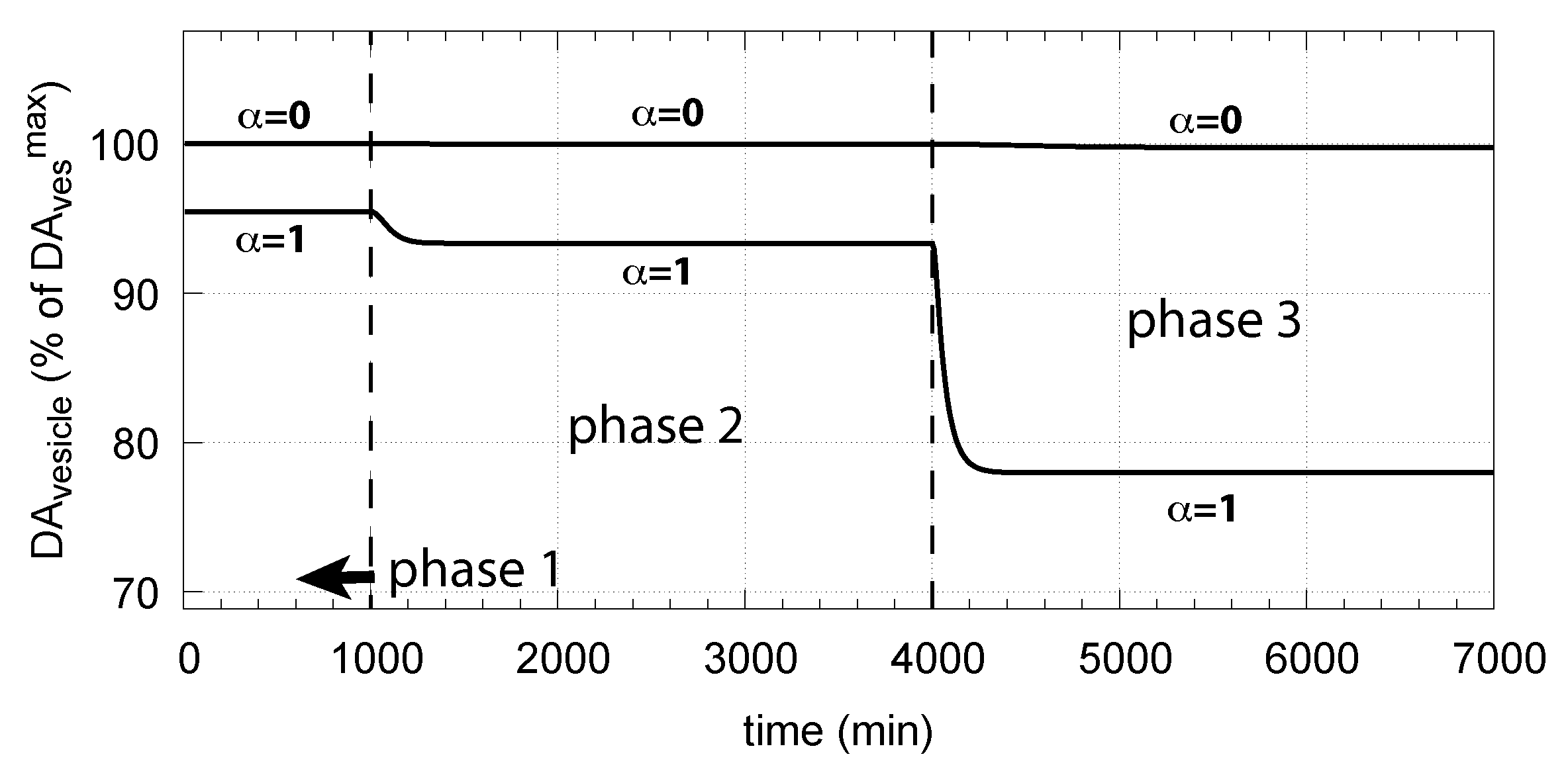
- When applied such that the combined fluxes + + are lower than ( is the rate of DA loading of vesicles, Figure 5), DOPA inflow helps to maintain DOPA homeostasis and slightly improves the performance of the controller/negative feedback. However, the improvement by DOPA addition is dependent on the controller accuracy (i.e., values). This is shown in Figure 10 where controller performances in absence of DOPA addition (panels a and b), and in its presence, (panels c and d) are compared for two different values. When is low and controller accuracy is high, DOPA addition improves controller performance and raises DA levels. In this case, all components of the model, including DA, are in a steady state.
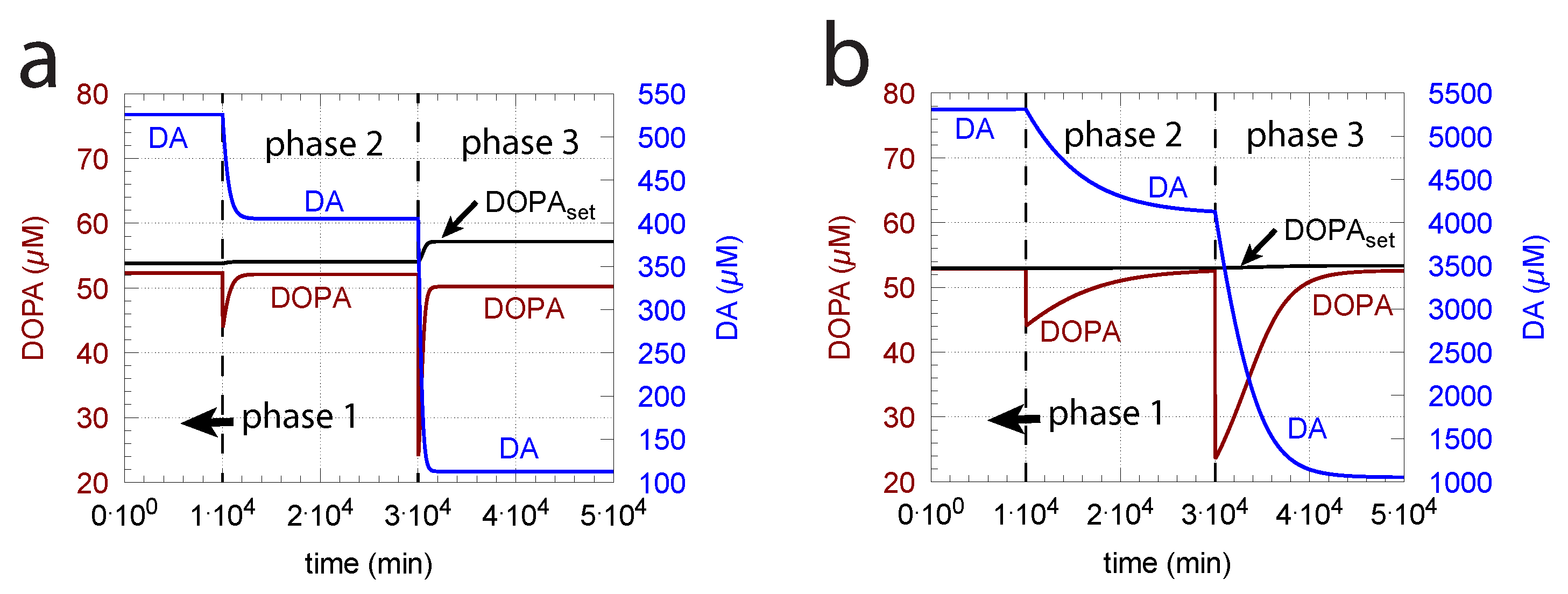
- (ii)
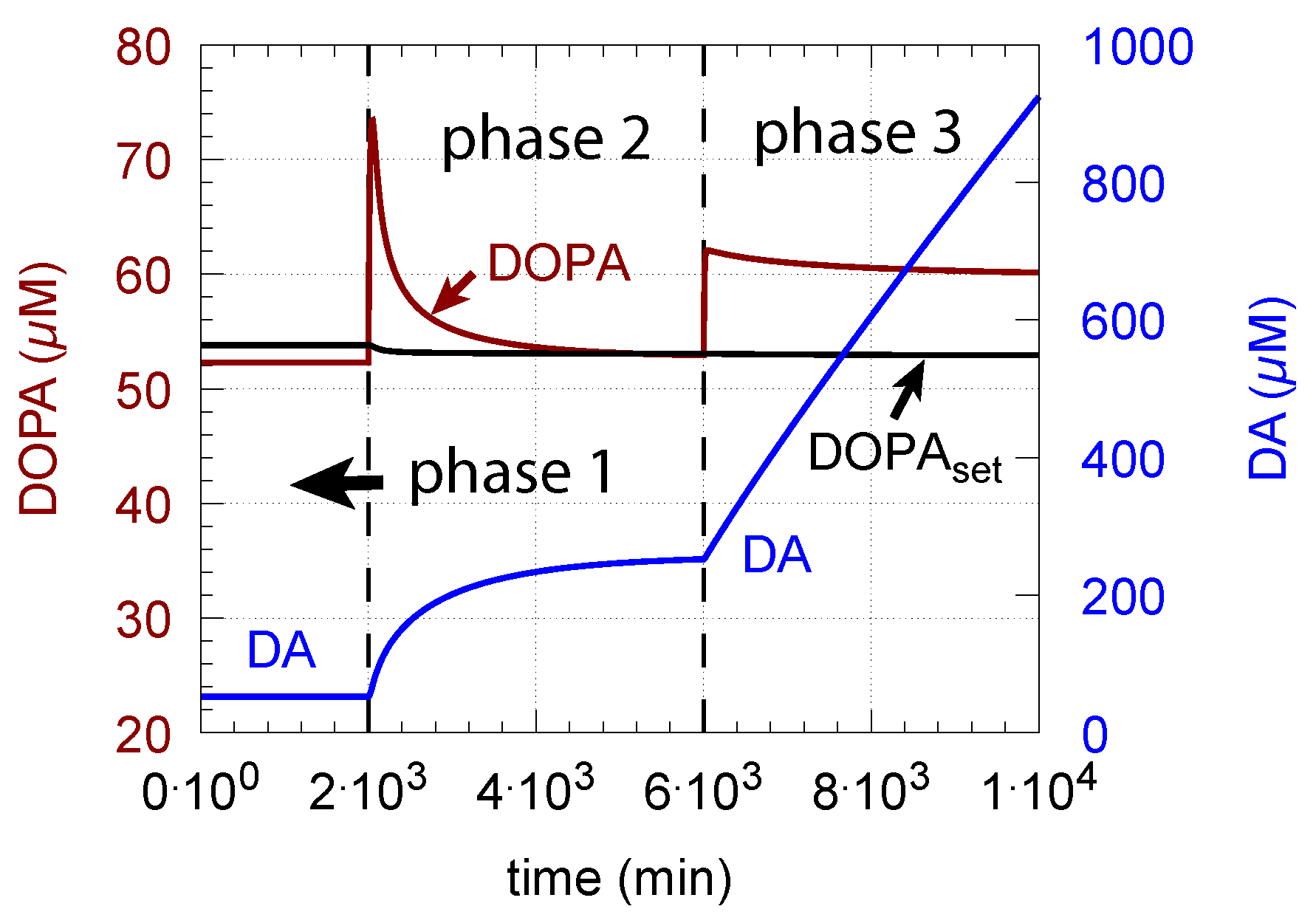
- However, if DOPA inflow results in the flux condition , then the controller breaks down, and DA levels start to grow continuously with a DOPA steady-state level above . The controller tries to oppose the increased DOPA levels by downregulating the compensatory flux to zero with a continuous increase in DA. This behavior, also termed integral wind-up [57], is shown in Figure 11, when the DOPA inflow rate in phase 3 was increased to = 8 M/min. DOPA steady-state levels are now entirely uncontrolled. Since the compensatory flux is practically zero, the steady-state level of DOPA is now solely determined by the DOPA inflow rate () and by the rates of DOPA removal.
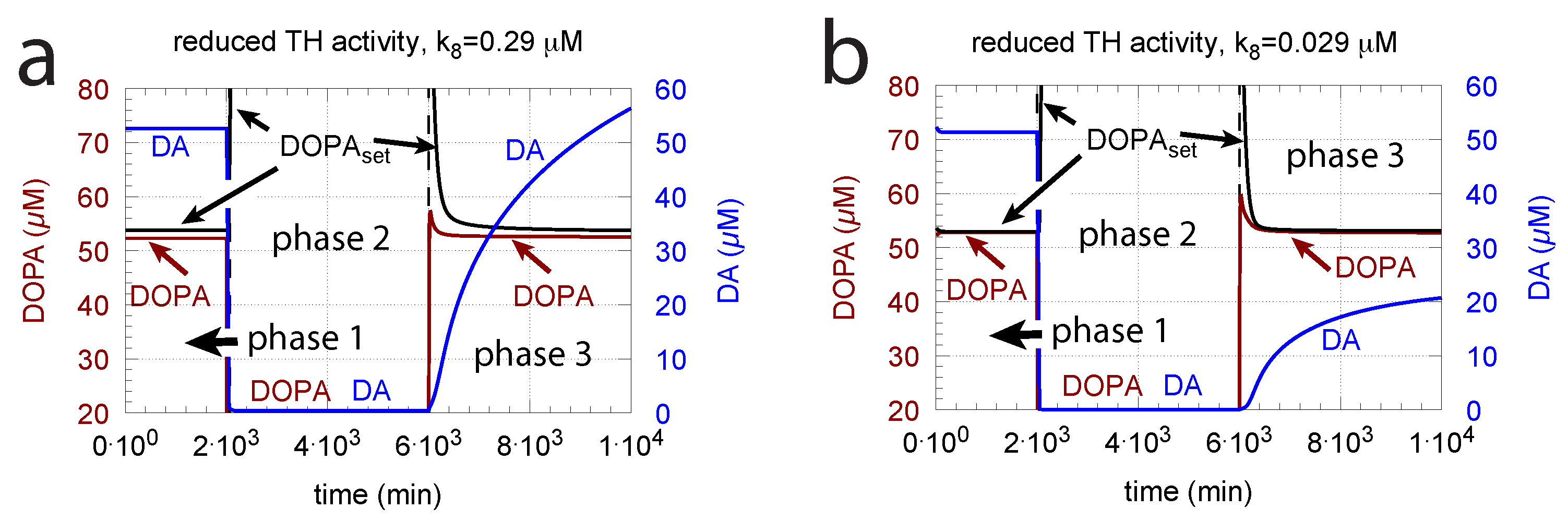
3.1.5. Robust DOPA Homeostasis Implies Maximum Vesicular DA Loading
3.1.6. Deteriorated DOPA Homeostasis by DA Removal/Auto-Oxidation
4. Discussion
4.1. On DOPA Regulation by DA Derepression in Cells
4.2. Why DOPA Homeostasis?
4.3. Oxidative Stress and Age
4.4. Why Only DOPA Inflow Control?
4.5. Role of Other TH Regulators
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| DA | dopamine |
| DAves | vesicular dopamine |
| DAex | extracellular dopamine |
| DAT | dopamine transporter |
| DDC | DOPA dehydroxylase |
| DOPA | L-3,4-dihydroxyphenylalanine |
| Levodopa | L-3,4-dihydroxyphenylalanine |
| MAO | monoamine oxidase |
| ROS | reactive oxygen species |
| t | time (min) |
| Tyr | tyrosine |
| TYR | tyrosinase |
| TH | tyrosine hydroxylase |
| VMAT2 | dopamine transporter located at vesicle membrane |
Appendix A. Derivation of Equation (5)
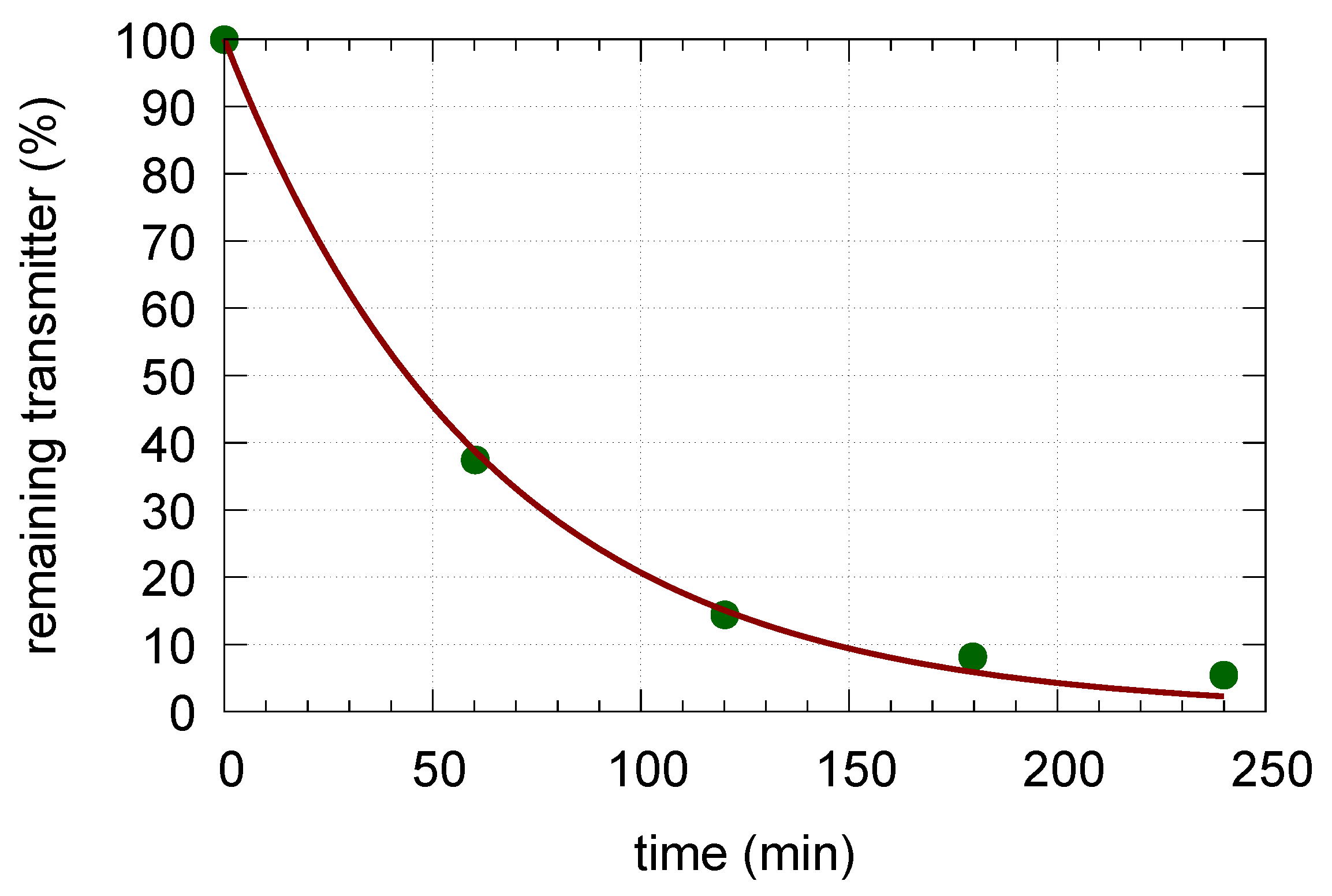
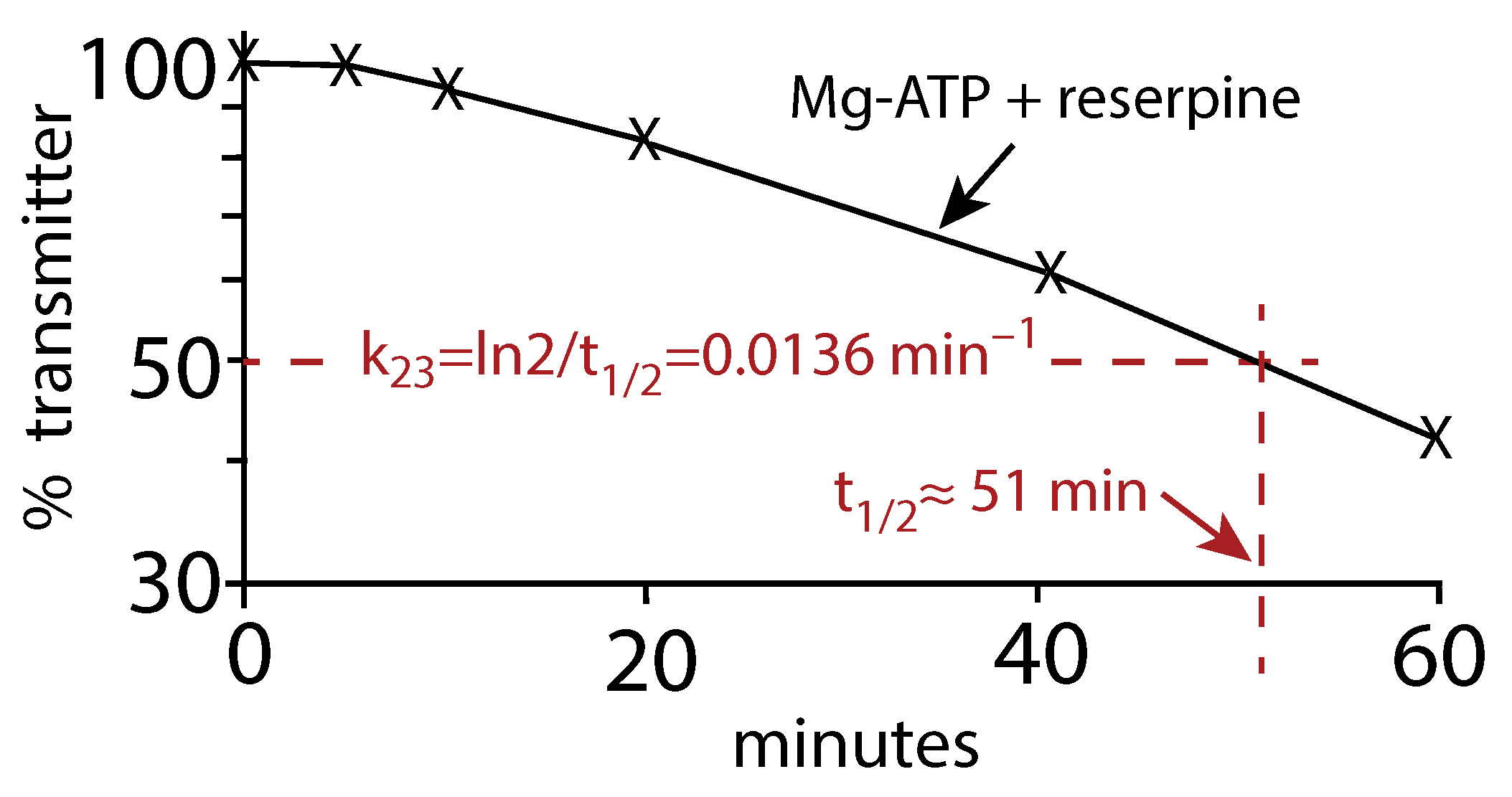
Appendix B. Determination of Rate Constant k23


References
- Wise, R.A.; Robble, M.A. Dopamine and addiction. Annu. Rev. Psychol. 2020, 71, 79–106. [Google Scholar] [CrossRef]
- Davis, K.L.; Kahn, R.S.; Ko, G.; Davidson, M. Dopamine in schizophrenia: A review and reconceptualization. Am. J. Psychiatry 1991, 148, 1474–1486. [Google Scholar]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Nieoullon, A. Dopamine and the regulation of cognition and attention. Prog. Neurobiol. 2002, 67, 53–83. [Google Scholar] [CrossRef]
- Friedman, M.H. Principles and Models of Biological Transport; Springer Science & Business Media: New York, NY, USA, 2008. [Google Scholar]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M.; Siegelbaum, S.; Hudspeth, A.J.; Mack, S. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2000; Volume 4. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-VCH Verlag: Weinheim, Germany, 2012. [Google Scholar]
- Bowling, K.M.; Huang, Z.; Xu, D.; Ferdousy, F.; Funderburk, C.D.; Karnik, N.; Neckameyer, W.; O’Donnell, J.M. Direct binding of GTP cyclohydrolase and tyrosine hydroxylase: Regulatory interactions between key enzymes in dopamine biosynthesis. J. Biol. Chem. 2008, 283, 31449–31459. [Google Scholar] [CrossRef] [Green Version]
- Cartier, E.A.; Parra, L.A.; Baust, T.B.; Quiroz, M.; Salazar, G.; Faundez, V.; Egaña, L.; Torres, G.E. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J. Biol. Chem. 2010, 285, 1957–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Park. Dis. 2012, 2012, 920953. [Google Scholar] [CrossRef]
- Chen, R.; Wei, J.; Fowler, S.C.; Wu, J.Y. Demonstration of functional coupling between dopamine synthesis and its packaging into synaptic vesicles. J. Biomed. Sci. 2003, 10, 774–781. [Google Scholar] [CrossRef]
- Jorge-Finnigan, A.; Kleppe, R.; Jung-Kc, K.; Ying, M.; Marie, M.; Rios-Mondragon, I.; Salvatore, M.F.; Saraste, J.; Martinez, A. Phosphorylation at serine 31 targets tyrosine hydroxylase to vesicles for transport along microtubules. J. Biol. Chem. 2017, 292, 14092–14107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvihill, K.G. Presynaptic regulation of dopamine release: Role of the DAT and VMAT2 transporters. Neurochem. Int. 2019, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Wayment, H.K.; Schenk, J.O.; Sorg, B.A. Characterization of extracellular dopamine clearance in the medial prefrontal cortex: Role of monoamine uptake and monoamine oxidase inhibition. J. Neurosci. 2001, 21, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Cragg, S.J.; Rice, M.E. Striatal dopamine neurotransmission: Regulation of release and uptake. Basal Ganglia 2016, 6, 123–148. [Google Scholar] [CrossRef] [Green Version]
- Fried, G. Noradrenaline release and uptake in isolated small dense cored vesicles from rat seminal ducts. Acta Physiol. Scand. 1981, 112, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Schonn, J.S.; Desnos, C.; Henry, J.P.; Darchen, F. Transmitter uptake and release in PC12 cells overexpressing plasma membrane monoamine transporters. J. Neurochem. 2003, 84, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.J. A small dopamine permeability of storage vesicle membranes and end product inhibition of tyrosine hydroxylase are sufficient to explain changes occurring in dopamine synthesis and storage after inhibition of neuron firing. Synapse 2007, 61, 715–723. [Google Scholar] [CrossRef]
- Hondebrink, L.; Meulenbelt, J.; Timmerman, J.G.; Van Den Berg, M.; Westerink, R.H. Amphetamine reduces vesicular dopamine content in dexamethasone-differentiated PC12 cells only following l-DOPA exposure. J. Neurochem. 2009, 111, 624–633. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Lazar, M.A.; Lockfeld, A.J.; Truscott, R.J.; Barchas, J.D. Tyrosine hydroxylase from bovine striatum: Catalytic properties of the phosphorylated and nonphosphorylated forms of the purified enzyme. J. Neurochem. 1982, 39, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Sura, G.R.; Daubner, S.C.; Fitzpatrick, P.F. Effects of phosphorylation by protein kinase A on binding of catecholamines to the human tyrosine hydroxylase isoforms. J. Neurochem. 2004, 90, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Royo, M.; Daubner, S.C.; Fitzpatrick, P.F. Effects of mutations in tyrosine hydroxylase associated with progressive dystonia on the activity and stability of the protein. Proteins 2005, 58, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Fossbakk, A.; Kleppe, R.; Knappskog, P.M.; Martinez, A.; Haavik, J. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in Dopa-responsive dystonia. Hum. Mutat. 2014, 35, 880–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, C.P. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 2014, 282, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, J.A.; Nijhout, H.F.; Reed, M.C. Homeostatic mechanisms in dopamine synthesis and release: A mathematical model. Theor. Biol. Med. Model. 2009, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogal, S.; Carotti, M.; Giaretta, L.; Lanciai, F.; Nogara, L.; Bubacco, L.; Bergantino, E. Human tyrosinase produced in insect cells: A landmark for the screening of new drugs addressing its activity. Mol. Biotechnol. 2015, 57, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Y.; Lee, J.R.; Xu, W.A.; Hahn, M.J.; Yang, J.M.; Park, Y.D. Effect of Cl− on tyrosinase: Complex inhibition kinetics and biochemical implication. J. Biomol. Struct. Dyn. 2007, 25, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Miller, G.W.; Voit, E.O. Computational systems analysis of dopamine metabolism. PLoS ONE 2008, 3, e2444. [Google Scholar] [CrossRef] [PubMed]
- Sura, G.R.; Lasagna, M.; Gawandi, V.; Reinhart, G.D.; Fitzpatrick, P.F. Effects of ligands on the mobility of an active-site loop in tyrosine hydroxylase as monitored by fluorescence anisotropy. Biochemistry 2006, 45, 9632–9638. [Google Scholar] [CrossRef] [Green Version]
- Quinsey, N.S.; Luong, A.Q.; Dickson, P.W. Mutational analysis of substrate inhibition in tyrosine hydroxylase. J. Neurochem. 1998, 71, 2132–2138. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Arthur Jr, R.E.; Thomas, D.M.; Elferink, L.A. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: Possible relevance to Parkinson’s disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef]
- Haavik, J. L-DOPA Is a Substrate for Tyrosine Hydroxylase. J. Neurochem. 1997, 69, 1720–1728. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, J.B.; Nicolaysen, L.C.; Michael, A.C. Modeling the dopaminergic nerve terminal. J. Neurosci. Methods 1988, 22, 239–252. [Google Scholar] [CrossRef]
- Kaushik, P.; Gorin, F.; Vali, S. Dynamics of tyrosine hydroxylase mediated regulation of dopamine synthesis. J. Comput. Neurosci. 2007, 22, 147–160. [Google Scholar] [CrossRef]
- Savageau, M.A. Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology; Addison-Wesley: Reading, MA, USA, 1976. [Google Scholar]
- Qi, Z.; Miller, G.W.; Voit, E.O. A mathematical model of presynaptic dopamine homeostasis: Implications for schizophrenia. Pharmacopsychiatry 2008, 41 (Suppl. S1), S89–S98. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Miller, G.W.; Voit, E.O. Computational modeling of synaptic neurotransmission as a tool for assessing dopamine hypotheses of schizophrenia. Pharmacopsychiatry 2010, 43 (Suppl. 1), S50–S60. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Wong-Lin, K. Integrated dopaminergic neuronal model with reduced intracellular processes and inhibitory autoreceptors. IET Syst. Biol. 2015, 9, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Véronneau-Veilleux, F.; Robaey, P.; Ursino, M.; Nekka, F. An integrative model of Parkinson’s disease treatment including levodopa pharmacokinetics, dopamine kinetics, basal ganglia neurotransmission and motor action throughout disease progression. J. Pharmacokinet. Pharmacodyn. 2021, 48, 133–148. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Mandali, A.; Chakravarthy, V.S.; Ramaswamy, S. A Computational Model of Loss of Dopaminergic Cells in Parkinson’s Disease Due to Glutamate-Induced Excitotoxicity. Front. Neural Circuits 2019, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Muddapu, V.R.; Chakravarthy, V.S. A Multi-Scale Computational Model of Excitotoxic Loss of Dopaminergic Cells in Parkinson’s Disease. Front. Neuroinform. 2020, 14, 34. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Vijayakumar, K.; Ramakrishnan, K.; Chakravarthy, V.S. A Computational Model of Levodopa-Induced Toxicity in Substantia Nigra Pars Compacta in Parkinson’s Disease. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Muddapu, V.R.; Chakravarthy, V.S. Influence of energy deficiency on the subcellular processes of Substantia Nigra Pars Compacta cell for understanding Parkinsonian neurodegeneration. Sci. Rep. 2021, 11, 1754. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B. Organization for Physiological Homeostasis. Physiol. Rev. 1929, 9, 399. [Google Scholar] [CrossRef]
- Langley, L.L. (Ed.) Homeostasis. Origins of the Concept; Dowden, Hutchinson & Ross, Inc.: Stroudsbourg, PA, USA, 1973. [Google Scholar]
- Ashby, W.R. An Introduction to Cybernetics; John Wiley: New York, NY, USA, 1956. [Google Scholar]
- Wiener, N. Cybernetics or Control and Communication in the Animal and the Machine; MIT Press: Cambridge, MA, USA, 1961. [Google Scholar]
- Cariani, P.A. The homeostat as embodiment of adaptive control. Int. J. Gen. Syst. 2009, 38, 139–154. [Google Scholar] [CrossRef]
- Kitano, H. Foundations of Systems Biology; The MIT Press: Cambridge, MA, USA; London, UK, 2001. [Google Scholar]
- Iglesias, P.A.; Ingalls, B.P. Control Theory and Systems Biology; MIT Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Ingalls, B.P. Mathematical Modeling in Systems Biology: An Introduction; MIT Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Hughes, G. (Ed.) Homeostasis and Feedback Mechanisms. In Symposia of the Society for Experimental Biology. Number XVIII; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Nijhout, H.F.; Best, J.A.; Reed, M.C. Systems biology of robustness and homeostatic mechanisms. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1440. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, J.; Johnson, M.A.; Katebi, R. Control Engineering: An Introductory Course; Palgrave Macmillan: Basingstoke, UK, 2002. [Google Scholar]
- Yi, T.M.; Huang, Y.; Simon, M.I.; Doyle, J. Robust perfect adaptation in bacterial chemotaxis through integral feedback control. Proc. Natl. Acad. Sci. USA 2000, 97, 4649–4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.M. Constructing Mathematical Models of Biological Signal Transduction Pathways: An Analysis of Robustness. In Foundations of Systems Biology; Kitano, H., Ed.; The MIT Press: Cambridge, MA, USA; London, UK, 2001; Chapter 12; pp. 251–260. [Google Scholar]
- Ni, X.Y.; Drengstig, T.; Ruoff, P. The control of the controller: Molecular mechanisms for robust perfect adaptation and temperature compensation. Biophys. J. 2009, 97, 1244–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolma, I.W.; Ni, X.Y.; Rensing, L.; Ruoff, P. Harmonic oscillations in homeostatic controllers: Dynamics of the p53 regulatory system. Biophys. J. 2010, 98, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, J.; Bagh, S.; Ingalls, B.P.; McMillen, D.R. Considerations for using integral feedback control to construct a perfectly adapting synthetic gene network. J. Theor. Biol. 2010, 266, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Drengstig, T.; Ruoff, P. Integrating fluctuating nitrate uptake and assimilation to robust homeostasis. Plant Cell Environ. 2012, 35, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Drengstig, T.; Jolma, I.W.; Ni, X.Y.; Thorsen, K.; Xu, X.M.; Ruoff, P. A basic set of homeostatic controller motifs. Biophys. J. 2012, 103, 2000–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briat, C.; Gupta, A.; Khammash, M. Antithetic Integral Feedback Ensures Robust Perfect Adaptation in Noisy Biomolecular Networks. Cell Syst. 2016, 2, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briat, C.; Zechner, C.; Khammash, M. Design of a Synthetic Integral Feedback Circuit: Dynamic Analysis and DNA Implementation. ACS Synth. Biol. 2016, 5, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Floros, I. Adaptive information processing of network modules to dynamic and spatial stimuli. BMC Syst. Biol. 2019, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Khammash, M.H. Perfect adaptation in biology. Cell Syst. 2021, 12, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.L. Applied Optimal Control & Estimation: Digital Design & Implementation; Prentice Hall: Englewood Cliffs, NJ, USA, 1992. [Google Scholar]
- Bennett, S. A brief history of automatic control. IEEE Control Syst. Mag. 1996, 16, 17–25. [Google Scholar] [CrossRef]
- Drengstig, T.; Ni, X.Y.; Thorsen, K.; Jolma, I.W.; Ruoff, P. Robust Adaptation and Homeostasis by Autocatalysis. J. Phys. Chem. B 2012, 116, 5355–5363. [Google Scholar] [CrossRef] [PubMed]
- Shoval, O.; Goentoro, L.; Hart, Y.; Mayo, A.; Sontag, E.; Alon, U. Fold-change detection and scalar symmetry of sensory input fields. Proc. Natl. Acad. Sci. USA 2010, 107, 15995–16000. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, K.; Hindmarsh, A.C. Description and Use of LSODE, the Livermore Solver for Ordinary Differential Equations. NASA Reference Publication 1327, Lawrence Livermore National Laboratory Report UCRL-ID-113855; National Aeronautics and Space Administration, Lewis Research Center: Cleveland, OH, USA, 1993.
- Volchegorskii, I.A.; Shemyakov, S.E.; Turygin, V.V.; Malinovskaya, N.V. The age dynamics of monoamine oxidase activity and levels of lipid peroxidation products in the human brain. Neurosci. Behav. Physiol. 2004, 34, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.A.; Esposito, G.; Enrico, P.; Mura, M.A.; Migheli, R.; Delogu, M.R.; Miele, M.; Desole, M.S.; Grella, G.; Miele, E. Manganese increases L-DOPA auto-oxidation in the striatum of the freely moving rat: Potential implications to L-DOPA long-term therapy of Parkinson’s disease. Br. J. Pharmacol. 2000, 130, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine autoxidation is controlled by acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.R.; Garris, P.A.; Kilts, C.D.; Wightman, R.M. Comparison of dopamine uptake in the basolateral amygdaloid nucleus, caudate-putamen, and nucleus accumbens of the rat. J. Neurochem. 1995, 64, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, Y.; Benoit-Marand, M.; Gonon, F.; Sulzer, D. Presynaptic regulation of dopaminergic neurotransmission. J. Neurochem. 2003, 87, 273–289. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyenbach, K.W.; Wieczorek, H. The V-type H+ ATPase: Molecular structure and function, physiological roles and regulation. J. Exp. Biol. 2006, 209, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Volz, T.J.; Hanson, G.R.; Fleckenstein, A.E. Measurement of kinetically resolved vesicular dopamine uptake and efflux using rotating disk electrode voltammetry. J. Neurosci. Methods 2006, 155, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Near, J.A. [3H]Dihydrotetrabenazine binding to bovine striatal synaptic vesicles. Mol. Pharmacol. 1986, 30, 252–257. [Google Scholar] [PubMed]
- Olefirowicz, T.M.; Ewing, A.G. Capillary electrophoresis in 2 and 5 microns diameter capillaries: Application to cytoplasmic analysis. Anal. Chem. 1990, 62, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Olefirowicz, T.M.; Ewing, A.G. Dopamine concentration in the cytoplasmic compartment of single neurons determined by capillary electrophoresis. J. Neurosci. Methods 1990, 34, 11–15. [Google Scholar] [CrossRef]
- Olefirowicz, T.M.; Ewing, A.G. capillary electrophoresis for sampling signal nerve cell. Chimia 1991, 45, 106–108. [Google Scholar]
- Chien, J.B.; Wallingford, R.A.; Ewing, A.G. Estimation of Free Dopamine in the Cytoplasm of the Giant Dopamine Cell of Planorbis corneus by Voltammetry and Capillary Electrophoresis. J. Neurochem. 1990, 54, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, M. Mammalian Dopa decarboxylase: Structure, catalytic activity and inhibition. Arch. Biochem. Biophys. 2014, 546, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, M.; Voltattorni, C.B. Multiple roles of the active site lysine of Dopa decarboxylase. Arch. Biochem. Biophys. 2009, 488, 130–139. [Google Scholar] [CrossRef]
- Martinez, A.; Abeygunawardana, C.; Haavik, J.; Flatmark, T.; Mildvan, A.S. Interaction of substrate and pterin cofactor with the metal of human tyrosine hydroxylase as determined by 1H-NMR. Adv. Exp. Med. Biol. 1993, 338, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Nasrin, S.; Ichinose, H.; Hidaka, H.; Nagatsu, T. Recombinant human tyrosine hydroxylase types 1-4 show regulatory kinetic properties for the natural (6R)-tetrahydrobiopterin cofactor. J. Biochem. 1994, 116, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Briggs, G.D.; Bulley, J.; Dickson, P.W. Catalytic domain surface residues mediating catecholamine inhibition in tyrosine hydroxylase. J. Biochem. 2014, 155, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Ichinose, H. Effect of metals and phenylalanine on the activity of human tryptophan hydroxylase-2: Comparison with that on tyrosine hydroxylase activity. Neurosci. Lett. 2006, 401, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, V.H., III; Walters, J.R.; Roth, R.H. Dopaminergic neurons—Alteration in the kinetic properties of tyrosine hydroxylase after cessation of impulse flow. Biochem. Pharmacol. 1976, 25, 655–661. [Google Scholar] [CrossRef]
- Li, M.; Hubálek, F.; Newton-Vinson, P.; Edmondson, D.E. High-level expression of human liver monoamine oxidase A in Pichia pastoris: Comparison with the enzyme expressed in Saccharomyces cerevisiae. Protein Expr. Purif. 2002, 24, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Papandreou, A.; Heales, S.J.; Kurian, M.A. Monoamine neurotransmitter disorders—Clinical advances and future perspectives. Nat. Rev. Neurol. 2015, 11, 567–584. [Google Scholar] [CrossRef]
- Nygaard, T.G.; Marsden, C.; Duvoisin, R. Dopa-responsive dystonia. Adv. Neurol. 1988, 50, 377–384. [Google Scholar]
- Wijemanne, S.; Jankovic, J. Dopa-responsive dystonia—Clinical and genetic heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Samardzic, K.; Raymond, B.B.A.; Djordjevic, S.P.; Rodgers, K.J. L-DOPA causes mitochondrial dysfunction in vitro: A novel mechanism of L-DOPA toxicity uncovered. Int. J. Biochem. Cell Biol. 2019, 117, 105624. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, P.; Delcambre, S.; Hanke, J.; Geffers, R.; Leist, M.; Hiller, K. Impairment of neuronal mitochondrial function by l-DOPA in the absence of oxygen-dependent auto-oxidation and oxidative cell damage. Cell Death Discov. 2021, 7, 151. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Sharabi, Y. Comparison of monoamine oxidase inhibitors in decreasing production of the autotoxic dopamine metabolite 3, 4-dihydroxyphenylacetaldehyde in PC12 cells. J. Pharmacol. Exp. Ther. 2016, 356, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Obata, T. Toward an evaluation of metabolite channeling in vivo. Curr. Opin. Biotechnol. 2020, 64, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef]
- Melov, S. Modeling mitochondrial function in aging neurons. Trends Neurosci. 2004, 27, 601–606. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I.; Selkoe, D.J.; Kelly, J.W. Protein Homeostasis; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011. [Google Scholar]
- Chaudhari, S.N.; Kipreos, E.T. The Energy Maintenance Theory of Aging: Maintaining Energy Metabolism to Allow Longevity. Bioessays 2018, 40, e1800005. [Google Scholar] [CrossRef] [PubMed]
- Drobac, G.; Waheed, Q.; Heidari, B.; Ruoff, P. An amplified derepression controller with multisite inhibition and positive feedback. PLoS ONE 2021, 16, e0241654. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, K.; Agafonov, O.; Selsto, C.H.; Jolma, I.W.; Ni, X.Y.; Drengstig, T.; Ruoff, P. Robust concentration and frequency control in oscillatory homeostats. PLoS ONE 2014, 9, e107766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleppe, R.; Waheed, Q.; Ruoff, P. DOPA Homeostasis by Dopamine: A Control-Theoretic View. Int. J. Mol. Sci. 2021, 22, 12862. https://doi.org/10.3390/ijms222312862
Kleppe R, Waheed Q, Ruoff P. DOPA Homeostasis by Dopamine: A Control-Theoretic View. International Journal of Molecular Sciences. 2021; 22(23):12862. https://doi.org/10.3390/ijms222312862
Chicago/Turabian StyleKleppe, Rune, Qaiser Waheed, and Peter Ruoff. 2021. "DOPA Homeostasis by Dopamine: A Control-Theoretic View" International Journal of Molecular Sciences 22, no. 23: 12862. https://doi.org/10.3390/ijms222312862
APA StyleKleppe, R., Waheed, Q., & Ruoff, P. (2021). DOPA Homeostasis by Dopamine: A Control-Theoretic View. International Journal of Molecular Sciences, 22(23), 12862. https://doi.org/10.3390/ijms222312862