
Disrupting the Molecular Pathway in Myotonic Dystrophy

{kind=link}
{kind=link}
{kind=link}
Abstract
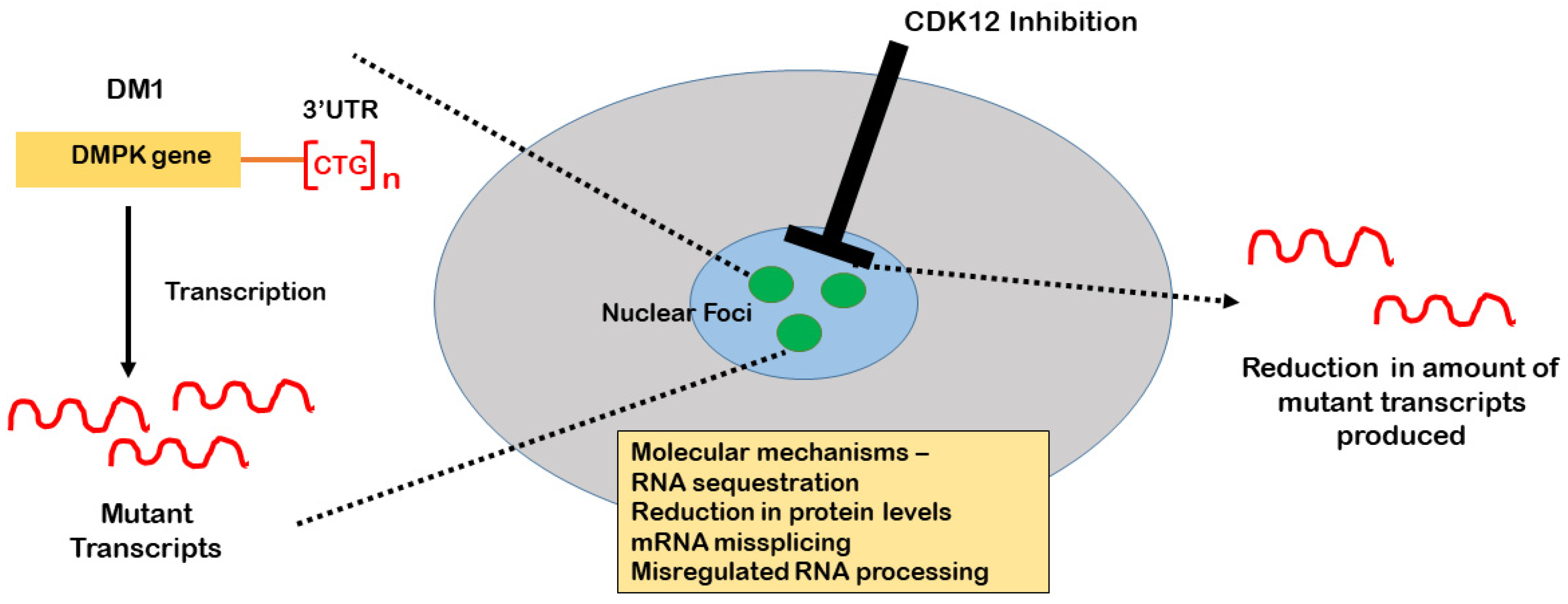
:1. Myotonic Dystrophy: Phenotype and Mutational Basis
2. The Role of CDK12 in the Production of DMPK Expansion Transcripts
3. CDK12 in RNA Processing
4. CDK12 as a Therapeutic Target for Myotonic Dystrophy
5. MBNL, CELF, and Other AS Factors Involved in DM Pathology
6. Domains and Sub-Cellular Localization Important for MBNL Function
7. Therapies to Alleviate MBNL Sequestration or Overexpress MBNL
8. Other Potential Therapeutic Interventions
9. RAN Translation
10. Conclusions
Funding
Conflicts of Interest
References
- Tapscott, S.J. Myotonic dystrophy, 3rd edition, by P. Harper, 436 pp., ill., London, W.B. Saunders, 2001, $85. Muscle Nerve 2002, 25, 926. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable ctg repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Ketley, A.; Wojciechowska, M.; Ghidelli-Disse, S.; Bamborough, P.; Ghosh, T.K.; Morato, M.L.; Sedehizadeh, S.; Malik, N.A.; Tang, Z.; Powalowska, P.; et al. CDK12 inhibition reduces abnormalities in cells from patients with myotonic dystrophy and in a mouse model. Sci. Transl. Med. 2020, 12, 541. [Google Scholar] [CrossRef]
- Ko, T.K.; Kelly, E.; Pines, J. CrkRS: A novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J. Cell Sci. 2001, 114 Pt 14, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohoutek, J.; Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Bösken, C.A.; Farnung, L.; Hintermair, C.; Schachter, M.M.; Vogel-Bachmayr, K.; Blazek, D.; Anand, K.; Fisher, R.P.; Eick, D.; Geyer, M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014, 5, 3505. [Google Scholar] [CrossRef] [Green Version]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, H.-C.; Lin, Y.; Chen, H.-R.; Fann, M.-J. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2016, 23, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Corden, J.L.; Cadena, D.L.; Ahearn, J.M.; Dahmus, M.E. A unique structure at the carboxyl terminus of the largest subunit of eukaryotic RNA polymerase II. Proc. Natl. Acad. Sci. USA 1985, 82, 7934–7938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, R.D.; Heidemann, M.; Hintermair, C.; Eick, D. Molecular evolution of the RNA polymerase II CTD. Trends Genet. 2008, 24, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Buratowski, S. The CTD code. Nat. Struct. Mol. Biol. 2003, 10, 679–680. [Google Scholar] [CrossRef]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Dubbury, S.J.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkowiak, B.; Greenleaf, A.L. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J. Biol. Chem. 2015, 290, 1786–1795. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Eick, D.; Bensaude, O. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 2013, 41, 1591–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Boyne, A.R.; Millhouse, S.R.; Manley, J.L. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65–Prp19 complex. Genes Dev. 2011, 25, 972–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, J.F.; Mazloomian, A.; Cheng, S.-W.G.; Hughes, C.S.; Chow, C.C.; Canapi, L.T.; Oloumi, A.; Trigo-Gonzalez, G.; Bashashati, A.; Xu, J.; et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017, 45, 6698–6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, L.; Muniz, L.; West, S. 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 2014, 28, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.T.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Wojciechowska, M.; Taylor, K.; Sobczak, K.; Napierala, M.; Krzyzosiak, W.J. Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 2014, 11, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek, M.O.; Jazurek, M.; Switonski, P.M.; Figura, G.; Krzyzosiak, W.J. Nuclear speckles are detention centers for transcripts containing expanded CAG repeats. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 1513–1520. [Google Scholar] [CrossRef]
- Holt, I.; Mittal, S.; Furling, D.; Butler-Browne, G.; Brook, J.D.; Morris, G.E. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells 2007, 12, 1035–1048. [Google Scholar] [CrossRef]
- Siboni, R.B.; Nakamori, M.; Wagner, S.D.; Struck, A.J.; Coonrod, L.A.; Harriott, S.A.; Cass, D.M.; Tanner, M.K.; Berglund, J.A. Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 2015, 13, 2386–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Jones, K.; Timchenko, N.A.; Timchenko, L. GSK3β is a new therapeutic target for myotonic dystrophy type 1. Rare Dis. 2013, 1, 4461–4472. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Weng, W.C.; Stock, L.; Lindquist, D.; Martines, A.; Gourdon, G.; Timchenko, N.; Snape, M.; Timchenko, L. Correction of Glycogen Synthase Kinase 3beta in Myotonic Dystrophy 1 Reduces the Mutant RNA and Improves Postnatal Survival of DMSXL Mice. Mol. Cell. Biol. 2019, 39, e00155-19. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3beta mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef] [Green Version]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmüller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Cooper, T.A. Roles for RNA-binding proteins in development and disease. Brain Res. 2016, 1647, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, M.; Swanson, M.S. RNA-binding protein misregulation in microsatellite expansion disorders. Adv. Exp. Med. Biol. 2014, 825, 353–388. [Google Scholar]
- Thomas, J.D.; Oliveira, R.; Sznajder, Ł.J.; Swanson, M.S. Myotonic dystrophy and developmental regulation of RNA processing. Compr. Physiol. 2011, 8, 509–553. [Google Scholar]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019, 28, 1312–1321. [Google Scholar] [CrossRef]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsawa, N.; Koebis, M.; Mitsuhashi, H.; Nishino, I.; Ishiura, S. ABLIM1 splicing is abnormal in skeletal muscle of patients with DM1 and regulated by MBNL, CELF and PTBP1. Genes Cells 2015, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Witten, J.T.; Ule, J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011, 27, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhuang, Y.; Batra, R.; Thomas, J.D.; Li, M.; Nutter, C.A.; Scotti, M.M.; Carter, H.A.; Wang, Z.J.; Huang, X.-S.; et al. HNRNPA1-induced spliceopathy in a transgenic mouse model of myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2020, 117, 5472–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, T.; Tuck, A.C.; Papasaikas, P.; Carl, S.H.; Flemr, M.; Knuckles, P.; Rankova, A.; Bühler, M.; Großhans, H. The RNA hairpin binder TRIM71 modulates alternative splicing by repressing MBNL1. Genes Dev. 2019, 33, 1221–1235. [Google Scholar] [CrossRef] [Green Version]
- Torres-Fernández, L.A.; Jux, B.; Bille, M.; Port, Y.; Schneider, K.; Geyer, M.; Mayer, G.; Kolanus, W. The mRNA repressor TRIM71 cooperates with Nonsense-Mediated Decay factors to destabilize the mRNA of CDKN1A/p21. Nucleic Acids Res. 2019, 47, 11861–11879. [Google Scholar] [CrossRef]
- Lavysh, D.; Neu-Yilik, G. UPF1-Mediated RNA Decay—Danse Macabre in a Cloud. Biomolecules 2020, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.M.D.A.; Tabach, Y.; Lourenco, G.; Armakola, M.; Ruvkun, G. Identification of genes in toxicity pathways of trinucleotide-repeat RNA in C. elegans. Nat. Struct. Mol. Biol. 2014, 21, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Teplova, M.; Patel, D.J. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol. Biol. 2008, 15, 1343–1351. [Google Scholar] [CrossRef]
- Hale, M.A.; Richardson, J.I.; Day, R.C.; McConnell, O.L.; Arboleda, J.; Wang, E.T.; Berglund, J.A. An engineered RNA binding protein with improved splicing regulation. Nucleic Acids Res. 2018, 46, 3152–3168. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Ramisetty, S.R.; Hussain, N.; Baranger, A.M. MBNL1-RNA recognition: Contributions of MBNL1 sequence and RNA conformation. ChemBioChem 2012, 13, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Grammatikakis, I.; Goo, Y.-H.; Echeverria, G.V.; Cooper, T.A. Identification of MBNL1 and MBNL3 domains required for splicing activation and repression. Nucleic Acids Res. 2011, 39, 2769–2780. [Google Scholar] [CrossRef]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Oma, Y.; Hattori, N.; Ishiura, S.; Nukina, N. Nuclear localization of MBNL1: Splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum. Mol. Genet. 2015, 24, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-Y.; Chang, K.-T.; Lin, Y.-M.; Kuo, T.-Y.; Wang, G.-S. Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 2018, 22, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Tabaglio, T.; Low, D.H.; Teo, W.K.L.; Goy, P.A.; Cywoniuk, P.; Wollmann, H.; Ho, J.; Tan, D.; Aw, J.; Pavesi, A.; et al. MBNL1 alternative splicing isoforms play opposing roles in cancer. Life Sci. Alliance 2018, 1, e201800157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Nakamori, M.; Matsumoto, J.; Murata, A.; Dohno, C.; Kiliszek, A.; Taylor, K.; Sobczak, K.; Nakatani, K. A Dimeric 2,9-Diamino-1,10-phenanthroline Derivative Improves Alternative Splicing in Myotonic Dystrophy Type 1 Cell and Mouse Models. Chemistry 2018, 24, 18115–18122. [Google Scholar] [CrossRef]
- Angelbello, A.J.; Benhamou, R.I.; Rzuczek, S.G.; Choudhary, S.; Tang, Z.; Chen, J.L.; Roy, M.; Wang, K.W.; Yildirim, I.; Jun, A.S.; et al. A Small Molecule that Binds an RNA Repeat Expansion Stimulates Its Decay via the Exosome Complex. Cell Chem. Biol. 2021, 28, 34–45.e6. [Google Scholar] [CrossRef]
- Lee, J.; Bai, Y.; Chembazhi, U.V.; Peng, S.; Yum, K.; Luu, L.M.; Hagler, L.D.; Serrano, J.F.; Chan, H.Y.E.; Kalsotra, A.; et al. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2019, 116, 8709–8714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Luu, L.M.; Peng, S.; Serrano, J.F.; Chan, E.; Zimmerman, S.C. Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J. Am. Chem. Soc. 2015, 137, 14180–14189. [Google Scholar] [CrossRef]
- Chakraborty, M.; Sellier, C.; Ney, M.; Villa, P.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Daunorubicin reduces MBNL1 sequestration caused by CUG-repeat expansion and rescues cardiac dysfunctions in a Drosophila model of myotonic dystrophy. Dis. Models Mech. 2018, 11, dmm032557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamori, M.; Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2016, 3, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-Y.; Guo, X.-M.; Wang, H.-Q.; Zhang, L.; Huang, S.-Y.; Huo, Y.-C.; Zhang, G.; Feng, J.-Z.; Zhang, R.-R.; Ma, Y.; et al. MBNL1 reverses the proliferation defect of skeletal muscle satellite cells in myotonic dystrophy type 1 by inhibiting autophagy via the mTOR pathway. Cell Death Dis. 2020, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Bargiela, A.; Sabater-Arcis, M.; Espinosa, J.P.E.; Zulaica, M.; de Munain, A.L.; Artero, R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA 2019, 116, 25203–25213. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Z.; Wu, M.; Zhang, L.; Wang, J.; Fu, W.; Xu, N.; Zhao, Z.; Lao, Y.; Xu, H. The natural compound neobractatin inhibits tumor metastasis by upregulating the RNA-binding-protein MBNL2. Cell Death Dis. 2019, 10, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, J.M.; Vaz, C.; Balaji, A.; Tanavde, V.; Talukdar, I. TNF-alpha regulates alternative splicing of genes participating in pathways of crucial metabolic syndromes; a transcriptome wide study. Cytokine 2020, 125, 154815. [Google Scholar] [CrossRef]
- Louis, J.M.; Agarwal, A.; Aduri, R.; Talukdar, I. Global analysis of RNA–protein interactions in TNF-α induced alternative splicing in metabolic disorders. FEBS Lett. 2021, 595, 476–490. [Google Scholar] [CrossRef]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-kappaB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef]
- Malakar, P.; Chartarifsky, L.; Hija, A.; Leibowitz, G.; Glaser, B.; Dor, Y.; Karni, R. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci. Rep. 2016, 6, 31222. [Google Scholar] [CrossRef]
- Altowyan, M.S.; Barakat, A.; Al-Majid, A.M.; Al-Ghulikah, H. Spiroindolone analogues bearing benzofuran moiety as a selective cyclooxygenase COX-1 with TNF-α and IL-6 inhibitors. Saudi J. Biol. Sci. 2020, 27, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Masuda, A.; Konishi, H.; Ohkawara, B.; Ito, M.; Kinoshita, M.; Kiyama, H.; Matsuura, T.; Ohno, K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci. Rep. 2016, 6, 25317. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Masuda, A.; Chen, G.; Bushra, S.; Kamon, M.; Araki, T.; Kinoshita, M.; Ohkawara, B.; Ito, M.; Ohno, K. Inhibition of cyclooxygenase-1 by nonsteroidal anti-inflammatory drugs demethylates MeR2 enhancer and promotes Mbnl1 transcription in myogenic cells. Sci. Rep. 2020, 10, 2558. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauvin, D.; Chrétien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; MacLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther.-Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, K.; Jenquin, J.R.; McConnell, O.L.; Cleary, J.D.; Richardson, J.I.; Pinto, B.S.; Haerle, M.C.; Delgado, E.; Planco, L.; Nakamori, M.; et al. A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. USA 2019, 116, 20991–21000. [Google Scholar] [CrossRef] [Green Version]
- Shokrollahi, M.; Mekhail, K. Interphase microtubules in nuclear organization and genome maintenance. Trends Cell Biol. 2021, 31, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Morato, M.; Brook, J.D. and Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front Neurol. 2018, 9, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, P.; Selma-Soriano, E.; Rapisarda, A.S.; Fernandez-Costa, J.M.; Perez-Alonso, M.; Artero, R. Myotonic dystrophy: Candidate small molecule therapeutics. Drug Discov. Today 2017, 22, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Thienes, C.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseley, M.L.; Zu, T.; Ikeda, Y.; Gao, W.; Mosemiller, A.K.; Daughters, R.S.; Chen, G.; Weatherspoon, M.R.; Clark, H.B.; Ebner, T.J.; et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006, 38, 758–769. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, T.; Ohtsuka, Y.; Okamura-Oho, Y.; Shikama, Y.; Yamada, M. Extended polyglutamine selectively interacts with caspase-8 and -10 in nuclear aggregates. Cell Death Differ. 2001, 8, 377–386. [Google Scholar]
- Zu, T.; Cleary, J.D.; Liu, Y.; Bañez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 2017, 95, 1292–1305.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, X.; Kumari, A.; Brown, J.; Brook, J.D. Disrupting the Molecular Pathway in Myotonic Dystrophy. Int. J. Mol. Sci. 2021, 22, 13225. https://doi.org/10.3390/ijms222413225
Xing X, Kumari A, Brown J, Brook JD. Disrupting the Molecular Pathway in Myotonic Dystrophy. International Journal of Molecular Sciences. 2021; 22(24):13225. https://doi.org/10.3390/ijms222413225
Chicago/Turabian StyleXing, Xiaomeng, Anjani Kumari, Jake Brown, and John David Brook. 2021. "Disrupting the Molecular Pathway in Myotonic Dystrophy" International Journal of Molecular Sciences 22, no. 24: 13225. https://doi.org/10.3390/ijms222413225
APA StyleXing, X., Kumari, A., Brown, J., & Brook, J. D. (2021). Disrupting the Molecular Pathway in Myotonic Dystrophy. International Journal of Molecular Sciences, 22(24), 13225. https://doi.org/10.3390/ijms222413225