
Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. CM[CAF-200/TPD] Promotes Resistance to Anti-HER2 Therapies in HER2-Positive Breast Cancer Cell Lines
2.2. Increased Expression of Epithelial–Mesenchymal Transition-Related Markers in Breast Cancer Cell Lines Was Induced by CM[CAF-200/TPD]
2.3. Changes in the Phosphorylation Pattern of HER2 and Downstream Signalling in Response to Anti-HER2 Therapies Was Induced by CM[CAF-200/TPD]
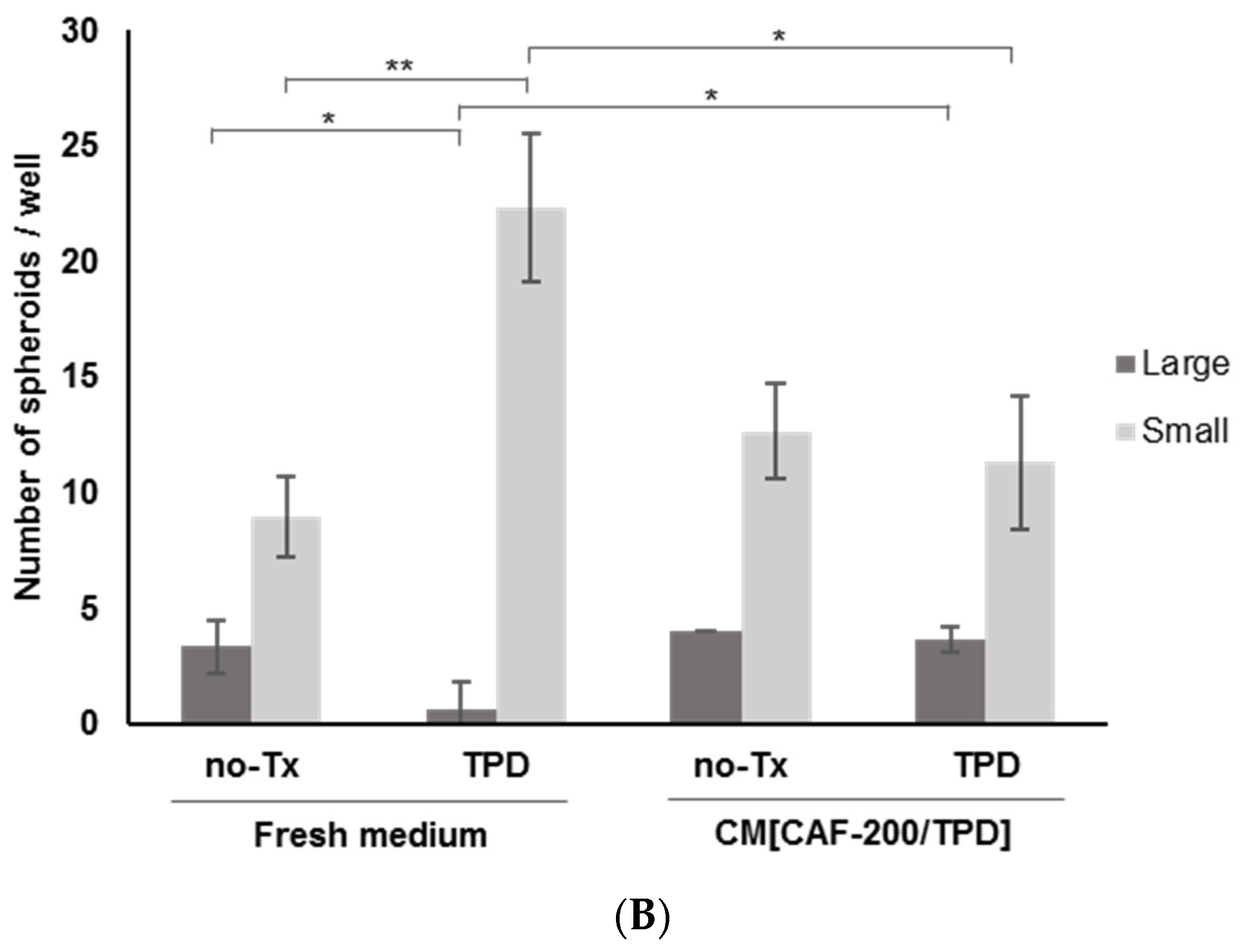
2.4. CAFs Induced a Spheroid-Forming Phenotype in BT-474 Cells Treated with Anti-HER2 Therapies Plus Chemotherapy
2.5. Tumour Cell Migration Increased in the Presence of the Molecular Milieu Secreted by CAFs
2.6. TME-Infiltrating miRNA-199b Could Be a Potential Target to Modulate Anti-HER2 Resistance in HER2-Positive BCCLs
2.7. Cytokine Secretion from CAF-200 Was Modified by Combined Treatment with TPD
2.8. Proteomic Analysis of CAF Secretome after Treatment Revealed Differentially Expressed Proteins
2.9. Gene Ontology and Functional Enrichment Analysis of CAF-200 Secretome Highlighted Oncogenic Processes and Regulation of Immune Response
2.10. Clinical Significance of the Different Protein Groupings
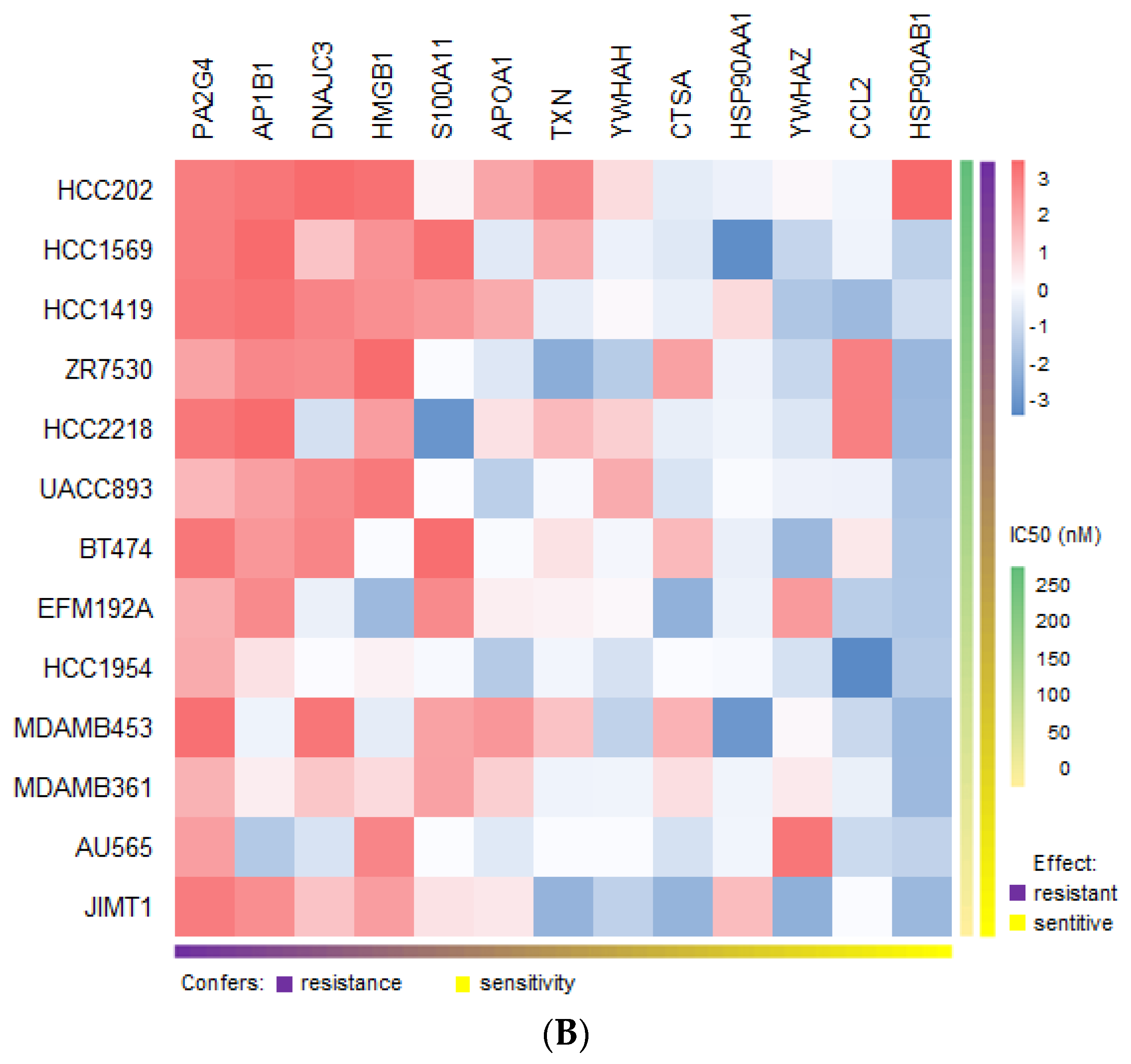
2.11. Suggested Role of the Resistance-Inducing Secretome in Drug Sensitivity and Resistance in HER2-Positive Breast Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Generation of Conditioned Medium (CM) Samples
4.3. Cell Proliferation Assays
4.4. Tumour Spheroid Formation Assay
4.5. Transwell Migration Assays
4.6. Protein Extraction and Quantification
4.7. Western Blotting (WB)
4.8. RNA Isolation
4.9. RNA Library Preparation and Sequencing
4.10. MiRNA Identification and Differential Expression Analysis
4.11. MiRNA-199b Transfection and Cell Proliferation Assay
4.12. Cytokine Arrays
4.13. Gene Ontology (GO) Analysis
4.14. Mass Spectrometry Analysis
4.15. Protein Identification and Quantification
4.16. Protein Data Analysis
4.17. Gene Set Enrichment Analysis
4.18. Kaplan-Meier Plotter Analysis
4.19. Analysis of Drug Sensitivity and Resistance
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the her-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.L.; Taghian, A.G.; Katz, M.S.; Niemierko, A.; Abi Raad, R.F.; Boon, W.L.; Bellon, J.R.; Wong, J.S.; Smith, B.L.; Harris, J.R. Breast cancer subtype approximated by estrogen receptor, progesterone receptor, and her-2 is associated with local and distant recurrence after breast-conserving therapy. J. Clin. Oncol. 2008, 26, 2373–2378. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pascual, T.; De Angelis, C.; Gutierrez, C.; Llombart-Cussac, A.; Wang, T.; Cortes, J.; Rexer, B.; Pare, L.; Forero, A.; et al. Her2-enriched subtype and erbb2 expression in her2-positive breast cancer treated with dual her2 blockade. J. Natl. Cancer Inst. 2020, 112, 46–54. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting erbb2 and discovering erbb3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on her2-positive human xenograft tumor models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in her2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Cortes, J.; Kim, S.B.; Im, S.A.; Hegg, R.; Im, Y.H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.C.; Ades, F.; de Azambuja, E.; Piccart-Gebhart, M. Trastuzumab for patients with her2 positive breast cancer: Delivery, duration and combination therapies. Breast 2013, 22 (Suppl. S2), S152–S155. [Google Scholar] [CrossRef]
- Aparicio, S.; Caldas, C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J.; Leyh, B. The impact of tumor stroma on drug response in breast cancer. Semin. Cancer Biol. 2015, 31, 3–15. [Google Scholar] [CrossRef]
- Howell, A.; Landberg, G.; Bergh, J. Breast tumour stroma is a prognostic indicator and target for therapy. Breast Cancer Res. 2009, 11 (Suppl. S3), S16. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403. [Google Scholar] [CrossRef] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M. Cancer-associated fibroblasts: A multifaceted driver of breast cancer progression. Cancer Lett. 2015, 361, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, Y.; Qu, Q.; Zhao, M.; Lou, Y.; Liu, J.; Huang, O.; Chen, X.; Wu, J.; Shen, K. Cancer-associated fibroblasts induce trastuzumab resistance in her2 positive breast cancer cells. Mol. Biosyst. 2015, 11, 1029–1040. [Google Scholar] [CrossRef]
- Fernandez-Nogueira, P.; Mancino, M.; Fuster, G.; Lopez-Plana, A.; Jauregui, P.; Almendro, V.; Enreig, E.; Menendez, S.; Rojo, F.; Noguera-Castells, A.; et al. Tumor-associated fibroblasts promote her2-targeted therapy resistance through fgfr2 activation. Clin. Cancer Res. 2020, 26, 1432–1448. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582. [Google Scholar] [CrossRef]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer associated fibroblasts: The architects of stroma remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. Microrna therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal micrornas that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. Microrna-181d-5p-containing exosomes derived from cafs promote emt by regulating cdx2/hoxa5 in breast cancer. Mol. Ther. Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Brohée, S.; Fumagalli, D.; Vincent, D.; Venet, D.; Ignatiadis, M.; Salgado, R.; Van den Eynden, G.; Rothé, F.; Desmedt, C.; et al. Constitutive phosphorylated stat3-associated gene signature is predictive for trastuzumab resistance in primary her2-positive breast cancer. BMC Med. 2015, 13, 177. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Zhao, Y.; Guo, B. Mir-199b-5p targets her2 in breast cancer cells. J. Cell. Biochem. 2013, 114, 1457–1463. [Google Scholar] [CrossRef]
- Hannafon, B.N.; Cai, A.; Calloway, C.L.; Xu, Y.-F.; Zhang, R.; Fung, K.-M.; Ding, W.-Q. Mir-23b and mir-27b are oncogenic micrornas in breast cancer: Evidence from a crispr/cas9 deletion study. BMC Cancer 2019, 19, 642. [Google Scholar] [CrossRef]
- Madoz-Gurpide, J.; Kuick, R.; Wang, H.; Misek, D.E.; Hanash, S.M. Integral protein microarrays for the identification of lung cancer antigens in sera that induce a humoral immune response. Mol. Cell. Proteom. 2008, 7, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Zazo, S.; Gonzalez-Alonso, P.; Martin-Aparicio, E.; Chamizo, C.; Cristobal, I.; Arpi, O.; Rovira, A.; Albanell, J.; Eroles, P.; Lluch, A.; et al. Generation, characterization, and maintenance of trastuzumab-resistant her2+ breast cancer cell lines. Am. J. Cancer Res. 2016, 6, 2661–2678. [Google Scholar]
- O’Brien, N.A.; Browne, B.C.; Chow, L.; Wang, Y.; Ginther, C.; Arboleda, J.; Duffy, M.J.; Crown, J.; O’Donovan, N.; Slamon, D.J. Activated phosphoinositide 3-kinase/akt signaling confers resistance to trastuzumab but not lapatinib. Mol. Cancer Ther. 2010, 9, 1489–1502. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin. Cancer Res. 2005, 11, 8425–8430. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Chen, D.; Huang, J.; Wang, R.; Feng, B.; Song, H.; Chen, L. Hmgb1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol. Cancer 2014, 13, 165. [Google Scholar] [CrossRef] [Green Version]
- Marín-Aguilera, M.; Codony-Servat, J.; Kalko, S.G.; Fernández, P.L.; Bermudo, R.; Buxo, E.; Ribal, M.J.; Gascón, P.; Mellado, B. Identification of docetaxel resistance genes in castration-resistant prostate cancer. Mol. Cancer Ther. 2012, 11, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Miles, D.; Kim, S.-B.; Im, Y.-H.; Im, S.-A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for her2-positive metastatic breast cancer (cleopatra): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (caf)-derived il32 promotes breast cancer cell invasion and metastasis via integrin beta3-p38 mapk signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; González, L.; Martínez-Ordoñez, A.; Fernandez-Garcia, B.; González, L.O.; Cid, S.; Dominguez, F.; Perez-Fernandez, R.; Vizoso, F.J. Cancer-associated fibroblasts affect breast cancer cell gene expression, invasion and angiogenesis. Cell Oncol. 2018, 41, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine tgf-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of emt and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer stem cell plasticity drives therapeutic resistance. Cancers 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-mesenchymal transition and breast cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Majewski, I.J.; Nuciforo, P.; Mittempergher, L.; Bosma, A.J.; Eidtmann, H.; Holmes, E.; Sotiriou, C.; Fumagalli, D.; Jimenez, J.; Aura, C.; et al. Pik3ca mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2–targeted therapies in breast cancer. J. Clin. Oncol. 2015, 33, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Wilks, S.T. Potential of overcoming resistance to her2-targeted therapies through the pi3k/akt/mtor pathway. Breast 2015, 24, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Jeong, A.J.; Ye, S.K. Highlighted stat3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, Q.; Gao, M.; Fu, L.; Li, Y.; Quan, H.; Lou, L. Stat3 activation confers trastuzumab-emtansine (t-dm1) resistance in her2-positive breast cancer. Cancer Sci. 2018, 109, 3305–3315. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of stat3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lin, B.W.; Chen, X.L.; Zhang, B.L.; Xiao, X.J.; Shi, J.S.; Lin, J.D.; Chen, X. Pai-1/pias3/stat3/mir-34a forms a positive feedback loop to promote emt-mediated metastasis through stat3 signaling in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Dagher, A.; Butterfield, C.; Moses, M.A. Adam12 is a novel regulator of tumor angiogenesis via stat3 signaling. Mol. Cancer Res. 2017, 15, 1608–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning stat3/5 by heat shock proteins: Interest of their targeting in cancer therapy. Cancers 2019, 12, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wang, F.B.; Li, Y.; Zeng, X.T. Down-regulation of mir-199b-5p is correlated with poor prognosis for breast cancer patients. Biomed. Pharmacother. 2016, 84, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yu, W.W.; Wang, L.L.; Peng, Y. Mir-130a acts as a potential diagnostic biomarker and promotes gastric cancer migration, invasion and proliferation by targeting runx3. Oncol. Rep. 2015, 34, 1153–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshar, E.; Hashemi-Arabi, M.; Salami, S.; Peirouvi, T.; Pouriran, R. Screening of acetaminophen-induced alterations in epithelial-to-mesenchymal transition-related expression of micrornas in a model of stem-like triple-negative breast cancer cells: The possible functional impacts. Gene 2019, 702, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Asukai, K.; Kawamoto, K.; Eguchi, H.; Konno, M.; Asai, A.; Iwagami, Y.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; et al. Micro-rna-130a-3p regulates gemcitabine resistance via pparg in cholangiocarcinoma. Ann. Surg. Oncol. 2017, 24, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, H.; Wang, Z.; Zhang, F.; Wei, H.; Li, L. Lncrna ccat1/mir-130a-3p axis increases cisplatin resistance in non-small-cell lung cancer cell line by targeting sox4. Cancer Biol. Ther. 2017, 18, 974–983. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, Y.; Qian, K.; Li, L.; Zhang, C.; Fu, X.; Zhang, X.; Chen, H.; Liu, Q.; Cao, S.; et al. A novel tumor suppressor zbtb1 regulates tamoxifen resistance and aerobic glycolysis through suppressing her2 expression in breast cancer. J. Biol. Chem. 2020, 295, 14140–14152. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, Z.; Sebzari, A.; Kordi-Tamandani, D.M.; Dastjerdi, K. Involvement of the dysregulation of mir-23b-3p, mir-195-5p, mir-656-5p, and mir-340-5p in trastuzumab resistance of her2-positive breast cancer cells and system biology approach to predict their targets involved in resistance. DNA Cell Biol. 2019, 38, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, K.; Resat, H. Constitutive activation of stat3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the il-6/jak/stat3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/jak/stat pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, Q.; Yu, H.; Wu, L.; Zhao, Y.; Zhang, C.; Yue, X.; Liu, Z.; Wu, H.; Haffty, B.G.; et al. Lif promotes tumorigenesis and metastasis of breast cancer through the akt-mtor pathway. Oncotarget 2014, 5, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C.; et al. The cc chemokine mcp-1/ccl2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar] [PubMed]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. Ccl2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. Ccl2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, P.W.; Rubin, J.S.; Miki, T.; Ron, D.; Aaronson, S.A. Human kgf is fgf-related with properties of a paracrine effector of epithelial cell growth. Science 1989, 245, 752–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Han, C.F.; Ni, T.; Di, L.; Liu, L.J.; Lv, W.C.; Bi, Y.R.; Jiang, N.; He, Y.; Li, H.M.; et al. A zeb1/p53 signaling axis in stromal fibroblasts promotes mammary epithelial tumours. Nat. Commun. 2019, 10, 3210. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The fgf family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamburger, A.W. The role of erbb3 and its binding partners in breast cancer progression and resistance to hormone and tyrosine kinase directed therapies. J. Mammary Gland. Biol. Neoplasia 2008, 13, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magadoux, L.; Isambert, N.; Plenchette, S.; Jeannin, J.F.; Laurens, V. Emerging targets to monitor and overcome docetaxel resistance in castration resistant prostate cancer (review). Int. J. Oncol. 2014, 45, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley, M.; Natarajan, S.K.; Mondal, J.; Best, D.; Goldman, D.; Shanthappa, B.; Pellowe, M.; Dash, C.; Saha, T.; Khiste, S.; et al. Nanoengineered disruption of heat shock protein 90 targets drug-induced resistance and relieves natural killer cell suppression in breast cancer. Cancer Res. 2020, 80, 5355–5366. [Google Scholar] [CrossRef]
- Sadeghirizi, A.; Yazdanparast, R.; Aghazadeh, S. Combating trastuzumab resistance by targeting thioredoxin-1/pten interaction. Tumour Biol. J. Int. Soc. Oncodevelop. Biol. Med. 2016, 37, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Yagoub, D.; Ahadi, A.; Nassif, N.T.; McGowan, E.M. Annexin/s100a protein family regulation through p14arf-p53 activation: A role in cell survival and predicting treatment outcomes in breast cancer. PLoS ONE 2017, 12, e0169925. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhan, Q.; Peng, X.; Qiu, Z.; Zhao, T. Ccl2 influences the sensitivity of lung cancer a549 cells to docetaxel. Oncol. Lett. 2018, 16, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, M.; Rosenberg, S.A. Tgf-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory cd8 t cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Ivy, P.; Timmer, W.; Khan, M.; Schulz, T.; Harris, P. Cancer-associated fibroblasts and therapies that interfere with their activity. Tumor Microenviron. Ther. 2013, 1, 19–36. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. Panther version 14: More genomes, a new panther go-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Rico-San Román, L.; Horcajo, P.; Regidor-Cerrillo, J.; Fernández-Escobar, M.; Collantes-Fernández, E.; Gutiérrez-Blázquez, D.; Hernáez-Sánchez, M.L.; Saeij, J.P.J.; Ortega-Mora, L.M. Comparative tachyzoite proteome analyses among six neospora caninum isolates with different virulence. Int. J. Parasitol. 2020, 50, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. Reporting recommendations for tumor marker prognostic studies. J. Clin. Oncol. 2005, 23, 9067–9072. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | logFC | p-Value |
|---|---|---|
| hsa-mir-130a-3p | 1.96 | 0.03 |
| hsa-let-7b-3p | 1.79 | 0.04 |
| hsa-mir-199b-5p | 1.76 | 0.03 |
| hsa-mir-4787-3p | −2.19 | 0.02 |
| hsa-mir-4281 | −2.04 | 0.04 |
| hsa-mir-4800-3p | −1.97 | 0.03 |
| hsa-mir-23b-3p | −1.95 | 0.02 |
| hsa-mir-4485-5p | −1.89 | 0.04 |
| hsa-mir-7854-3p | −1.79 | 0.03 |
| Cytokine | Intensity | p-Value | Differential Intensity | Relative Fold Change | log2 FC | # Hits |
|---|---|---|---|---|---|---|
| Angiogenin | 0.3199 | 0.0028 | −0.1331 | −0.2938 | −0.5019 | 5 |
| BDNF | 0.4531 | 0.4293 | −0.0297 | −0.0616 | −0.0917 | 1 |
| BLC (CXCL13) | 0.0715 | 0.1307 | −0.0895 | −0.5558 | −1.1708 | 0 |
| Ck beta 8-1 (CCL23) | 0.0322 | 0.1344 | −0.1055 | −0.7660 | −2.0955 | 0 |
| EGF | 0.0100 | 0.0904 | −0.1008 | −0.9097 | −3.4698 | 0 |
| ENA-78 (CXCL5) | 0.0464 | 0.1580 | −0.0459 | −0.4973 | −0.9922 | 0 |
| Eotaxin-1 (CCL11) | 0.0208 | 0.1268 | −0.0918 | −0.8155 | −2.4387 | 0 |
| Eotaxin-2 (CCL24) | 0.0100 | 0.1782 | −0.0778 | −0.8861 | −3.1341 | 0 |
| Eotaxin-3 (CCL26) | 0.0100 | 0.0456 | −0.0325 | −0.7647 | −2.0872 | 2 |
| FGF-4 | 0.1738 | 0.4402 | 0.0288 | 0.1987 | 0.2614 | 0 |
| FGF-6 | 0.1882 | 0.0561 | 0.0600 | 0.4679 | 0.5538 | 1 |
| FGF-7 (KGF) | 0.0997 | 0.1638 | 0.0436 | 0.7776 | 0.8300 | 4 |
| FGF-9 | 0.2474 | 0.5959 | 0.0315 | 0.1460 | 0.1966 | 1 |
| FLT-3 Ligand | 0.1239 | 0.5896 | −0.0123 | −0.0906 | −0.1370 | 0 |
| Fractalkine (CX3CL1) | 0.1396 | 0.1526 | −0.0589 | −0.2969 | −0.5082 | 0 |
| G-CSF | 0.0100 | 0.1813 | −0.0391 | −0.7962 | −2.2948 | 0 |
| GDNF | 0.0393 | 0.0590 | −0.1486 | −0.7907 | −2.2565 | 3 |
| GM-CSF | 0.0100 | 0.0739 | −0.0320 | −0.7621 | −2.0716 | 0 |
| GPC-2 (CXCL6) | 0.0466 | 0.0889 | −0.1052 | −0.6931 | −1.7041 | 0 |
| GRO a/b/g | 0.1577 | 0.0313 | −0.1014 | −0.3913 | −0.7162 | 2 |
| GRO alpha (CXCL1) | 0.0100 | 0.1253 | −0.0267 | −0.7279 | −1.8775 | 0 |
| HGF | 0.0100 | 0.1463 | −0.0363 | −0.7842 | −2.2124 | 0 |
| I-309 (CCL1) | 0.0338 | 0.5121 | −0.0289 | −0.4605 | −0.8904 | 0 |
| IFN-gamma | 0.1881 | 0.9113 | 0.0051 | 0.0281 | 0.0400 | 0 |
| IGF-1 | 0.0100 | 0.0384 | −0.0750 | −0.8824 | −3.0874 | 3 |
| IGFBP-1 | 0.0100 | 0.2575 | −0.0365 | −0.7851 | −2.2180 | 0 |
| IGFBP-2 | 0.0253 | 0.0948 | −0.0776 | −0.7542 | −2.0247 | 0 |
| IGFBP-3 | 0.1122 | 0.9345 | 0.0010 | 0.0092 | 0.0133 | 0 |
| IGFBP-4 | 0.1027 | 0.2169 | −0.0222 | −0.1774 | −0.2817 | 0 |
| IL-1 alpha (IL-1 F1) | 0.0770 | 0.8593 | −0.0083 | −0.0977 | −0.1483 | 0 |
| IL-1 beta (IL-1 F2) | 0.2268 | 0.3119 | 0.0318 | 0.1633 | 0.2182 | 0 |
| IL-10 | 0.0100 | NA | 0.0000 | 0.0000 | 0.0000 | 0 |
| IL-12 (p40/p70) | 0.1481 | 0.0983 | −0.0269 | −0.1536 | −0.2406 | 0 |
| IL-13 | 0.0654 | 0.3267 | 0.0323 | 0.9763 | 0.9828 | 1 |
| IL-15 | 0.2774 | 0.2844 | 0.0478 | 0.2081 | 0.2727 | 2 |
| IL-16 | 0.1368 | 0.2513 | −0.0505 | −0.2697 | −0.4535 | 0 |
| IL-2 | 0.1239 | 0.5033 | 0.0271 | 0.2799 | 0.3561 | 0 |
| IL-3 | 0.1964 | 0.6411 | 0.0224 | 0.1290 | 0.1751 | 0 |
| IL-4 | 0.0869 | 0.2814 | −0.0215 | −0.1984 | −0.3191 | 0 |
| IL-5 | 0.0100 | 0.1043 | −0.0978 | −0.9073 | −3.4306 | 3 |
| IL-6 | 1.8106 | 0.5854 | −0.0546 | −0.0293 | −0.0429 | 1 |
| IL-7 | 0.0100 | 0.0949 | −0.1245 | −0.9256 | −3.7491 | 4 |
| IL-8 (CXCL8) | 1.0915 | 0.9680 | 0.0061 | 0.0057 | 0.0082 | 1 |
| IP-10 (CXCL10) | 0.6526 | 0.2375 | 0.0728 | 0.1256 | 0.1707 | 4 |
| Leptin | 0.1577 | 0.2038 | −0.0463 | −0.2268 | −0.3711 | 0 |
| LIF | 0.3174 | 0.5290 | 0.0531 | 0.2011 | 0.2643 | 4 |
| LIGHT (TNFSF14) | 0.1716 | 0.7360 | −0.0059 | −0.0331 | −0.0486 | 0 |
| MCP-1 (CCL2) | 1.5040 | 0.1989 | 0.1576 | 0.1170 | 0.1596 | 4 |
| MCP-2 (CCL8) | 0.0707 | 0.0456 | −0.0596 | −0.4575 | −0.8824 | 1 |
| MCP-3 (CCL7) | 0.0100 | 0.0017 | −0.0681 | −0.8719 | −2.9649 | 2 |
| MCP-4 (CCL13) | 0.0676 | 0.1183 | −0.0606 | −0.4727 | −0.9232 | 0 |
| M-CSF | 0.0577 | 0.0341 | −0.1840 | −0.7613 | −2.0669 | 2 |
| MDC (CCL22) | 0.0100 | 0.0452 | −0.0986 | −0.9079 | −3.4404 | 1 |
| MIF | 0.0362 | 0.0042 | −0.1472 | −0.8025 | −2.3398 | 5 |
| MIG (CXCL9) | 0.0100 | 0.1174 | −0.0204 | −0.6708 | −1.6028 | 0 |
| MIP-1 beta (CCL4) | 0.2621 | 0.0575 | −0.0705 | −0.2120 | −0.3437 | 1 |
| MIP-1 delta | 0.0100 | 0.0006 | −0.0684 | −0.8725 | −2.9711 | 3 |
| MIP-3 alpha (CCL20) | 0.0100 | 0.0146 | −0.0516 | −0.8377 | −2.6232 | 1 |
| NAP-2 (CXCL7) | 0.0100 | 0.0856 | −0.0692 | −0.8737 | −2.9850 | 0 |
| NT-3 | 0.3264 | 0.3826 | −0.0320 | −0.0894 | −0.1351 | 1 |
| NT-4 | 0.0807 | 0.4584 | −0.0204 | −0.2020 | −0.3256 | 0 |
| OPG (TNFR SF 11) | 1.3666 | 0.6382 | 0.0410 | 0.0309 | 0.0439 | 1 |
| OPN (SSP1) | 0.3109 | 0.1552 | −0.0895 | −0.2235 | −0.3649 | 1 |
| OSM | 0.4078 | 0.6803 | 0.0178 | 0.0455 | 0.0642 | 1 |
| PARC | 0.1914 | 0.8600 | −0.0119 | −0.0586 | −0.0871 | 0 |
| PDGF-BB | 0.0948 | 0.3120 | −0.0265 | −0.2187 | −0.3561 | 0 |
| PLGF | 0.1465 | 0.3941 | 0.0627 | 0.7481 | 0.8058 | 0 |
| RANTES (CCL5) | 0.2646 | 0.6313 | 0.0288 | 0.1221 | 0.1662 | 1 |
| SCF | 0.1688 | 0.2936 | 0.0226 | 0.1546 | 0.2073 | 1 |
| SDF-1 alpha (CXCL12) | 0.2151 | 0.9245 | −0.0024 | −0.0112 | −0.0163 | 0 |
| TARC (CCL17) | 0.3281 | 0.5687 | −0.0430 | −0.1158 | −0.1775 | 1 |
| TGF beta 1 | 0.0544 | 0.0192 | −0.0718 | −0.5692 | −1.2149 | 1 |
| TGF beta 2 | 0.6476 | 0.0122 | 0.1226 | 0.2335 | 0.3028 | 5 |
| TGF beta 3 | 0.1224 | 0.3794 | −0.0171 | −0.1228 | −0.1890 | 0 |
| TIMP-1 | 0.6717 | 0.4984 | −0.0302 | −0.0430 | −0.0634 | 1 |
| TIMP-2 | 1.3389 | 0.1198 | −0.0474 | −0.0342 | −0.0502 | 1 |
| TNF alpha | 0.0100 | 0.0013 | −0.0729 | −0.8794 | −3.0519 | 3 |
| TNF beta (TNF SF 1B) | 0.0536 | 0.0996 | −0.0943 | −0.6377 | −1.4649 | 0 |
| TPO | 0.1176 | 0.0026 | 0.0337 | 0.4011 | 0.4865 | 4 |
| VEGF-A | 0.2579 | 0.6450 | 0.0224 | 0.0952 | 0.1312 | 1 |
| Pathway Name | Pathway Identifier | Proteins from Study Found in Pathway | no. Proteins in Study (Total) |
|---|---|---|---|
| Immune System | R-HSA-168256 | AP1B1, CCL2, CTSA, DNAJC3, HMGB1, HSP90AA1, HSP90AB1, PA2G4, S100A11, TXN, YWHAZ | 11 (2895) |
| Innate Immune System | R-HSA-168249 | CTSA, DNAJC3, HMGB1, HSP90AA1, HSP90AB1, PA2G4, S100A11, TXN | 8 (1331) |
| Neutrophil degranulation | R-HSA-6798695 | CTSA, DNAJC3, HMGB1, HSP90AA1, HSP90AB1, PA2G4, S100A11 | 7 (480) |
| Disease | R-HSA-1643685 | AP1B1, APOA1, CTSA, DNAJC3, HSP90AA1, HSP90AB1, TXN | 7 (2512) |
| Signal Transduction | R-HSA-162582 | APOA1, CCL2, HSP90AA1, HSP90AB1, SERPINE1, YWHAH, YWHAZ | 7 (3421) |
| Metabolism of proteins | R-HSA-392499 | APOA1, CCL2, CTSA, DNAJC3, EIF3F, TXN | 6 (2355) |
| Vesicle-mediated transport | R-HSA-5653656 | AP1B1, APOA1, HSP90AA1, YWHAH, YWHAZ | 5 (825) |
| Infectious disease | R-HSA-5663205 | AP1B1, DNAJC3, HSP90AA1, HSP90AB1, TXN | 5 (1468) |
| Gene expression (Transcription) | R-HSA-74160 | HSP90AA1, SERPINE1, TXN, YWHAH, YWHAZ | 5 (1851) |
| Metabolism | R-HSA-1430728 | APOA1, CTSA, HSP90AA1, HSP90AB1, TXN | 5 (3658) |
| Programmed Cell Death | R-HSA-5357801 | HMGB1, HSP90AA1, YWHAH, YWHAZ | 4 (218) |
| Cell Cycle | R-HSA-1640170 | HSP90AA1, HSP90AB1, YWHAH, YWHAZ | 4 (734) |
| Signalling by Interleukins | R-HSA-449147 | CCL2, HMGB1, HSP90AA1, YWHAZ | 4 (647) |
| Cytokine Signalling in Immune system | R-HSA-1280215 | CCL2, HMGB1, HSP90AA1, YWHAZ | 4 (1332) |
| Generic Transcription Pathway | R-HSA-212436 | SERPINE1, TXN, YWHAH, YWHAZ | 4 (1554) |
| RNA Polymerase II Transcription | R-HSA-73857 | SERPINE1, TXN, YWHAH, YWHAZ | 4 (1693) |
| TP53 Regulates Metabolic Genes | R-HSA-5628897 | TXN, YWHAH, YWHAZ | 3 (125) |
| HSP90 chaperone cycle for steroid hormone receptors (SHR) | R-HSA-3371497 | HSP90AA1, HSP90AB1 | 2 (80) |
| Resistance of ERBB2 KD mutants to trastuzumab | R-HSA-9665233 | HSP90AA1 | 1 (5) |
| Drug resistance in ERBB2 TMD/JMD mutants | R-HSA-9665737 | HSP90AA1 | 1 (5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luque, M.; Sanz-Álvarez, M.; Santamaría, A.; Zazo, S.; Cristóbal, I.; de la Fuente, L.; Mínguez, P.; Eroles, P.; Rovira, A.; Albanell, J.; et al. Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 13297. https://doi.org/10.3390/ijms222413297
Luque M, Sanz-Álvarez M, Santamaría A, Zazo S, Cristóbal I, de la Fuente L, Mínguez P, Eroles P, Rovira A, Albanell J, et al. Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer. International Journal of Molecular Sciences. 2021; 22(24):13297. https://doi.org/10.3390/ijms222413297
Chicago/Turabian StyleLuque, Melani, Marta Sanz-Álvarez, Andrea Santamaría, Sandra Zazo, Ion Cristóbal, Lorena de la Fuente, Pablo Mínguez, Pilar Eroles, Ana Rovira, Joan Albanell, and et al. 2021. "Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer" International Journal of Molecular Sciences 22, no. 24: 13297. https://doi.org/10.3390/ijms222413297
APA StyleLuque, M., Sanz-Álvarez, M., Santamaría, A., Zazo, S., Cristóbal, I., de la Fuente, L., Mínguez, P., Eroles, P., Rovira, A., Albanell, J., Madoz-Gúrpide, J., & Rojo, F. (2021). Targeted Therapy Modulates the Secretome of Cancer-Associated Fibroblasts to Induce Resistance in HER2-Positive Breast Cancer. International Journal of Molecular Sciences, 22(24), 13297. https://doi.org/10.3390/ijms222413297