
Targeted Long-Read Sequencing Decodes the Transcriptional Atlas of the Founding RAS Gene Family Members



Abstract
:1. Introduction
2. Results
2.1. Nanopore Sequencing Reveals Novel RAS mRNA Transcripts
2.2. Identification of Novel KRAS mRNA Transcripts
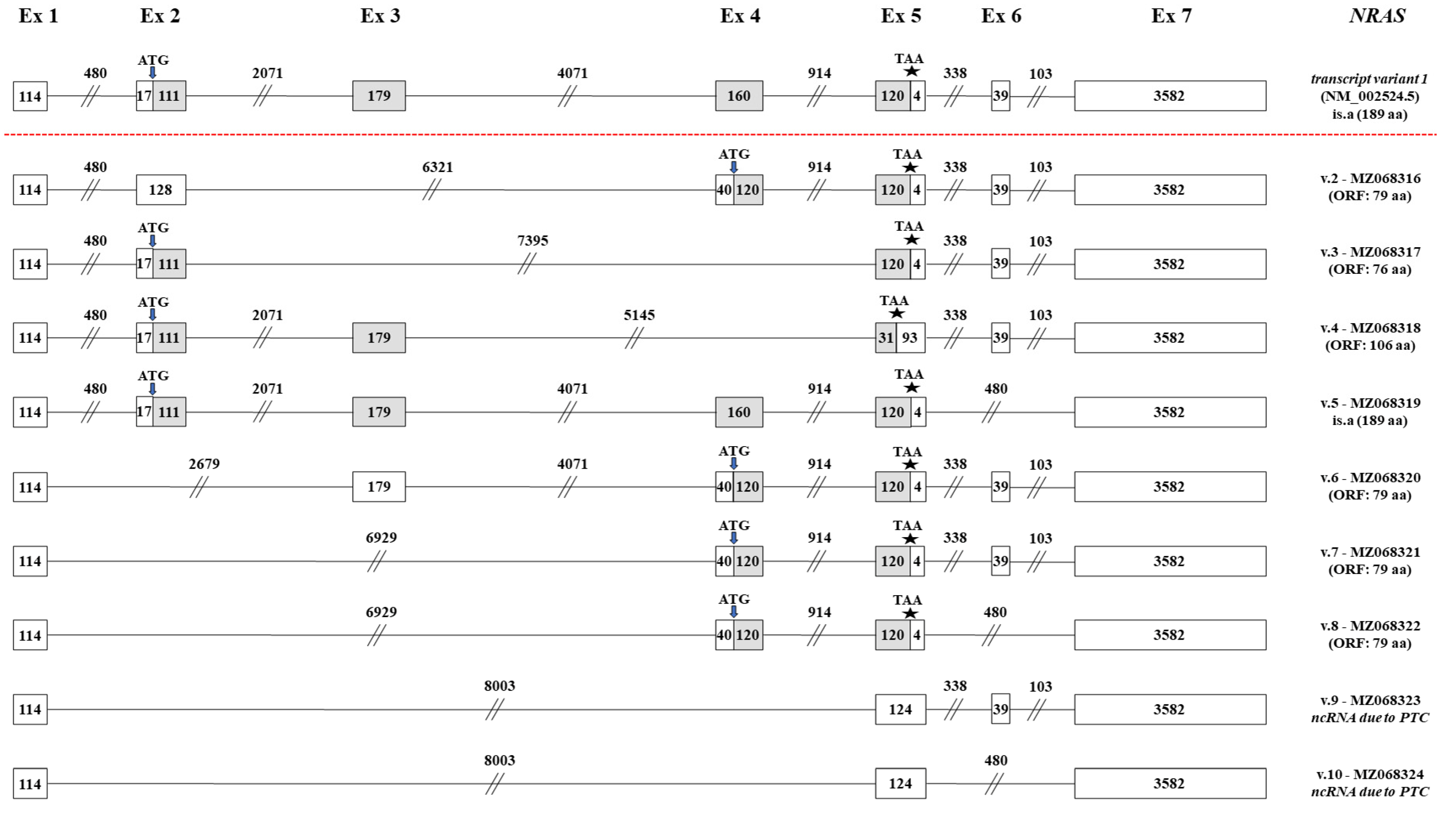
2.3. Identification of Novel NRAS mRNA Transcripts
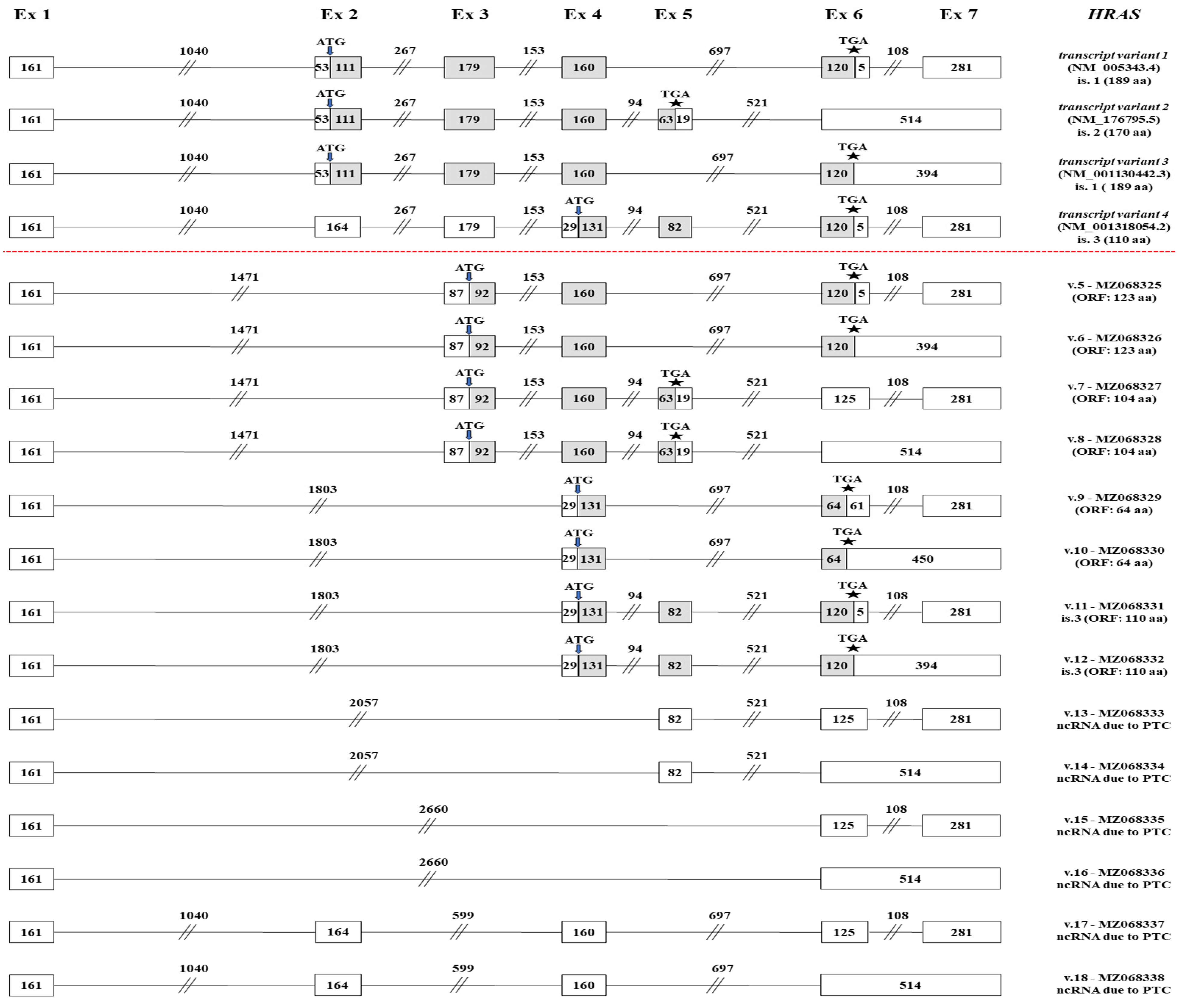
2.4. Identification of Novel HRAS mRNA Transcripts
2.5. Expression Pattern of the Novel RAS Transcripts in a Wide Spectrum of Human Tissues
3. Discussion
4. Materials and Methods
4.1. Culture of Human Cell Lines
4.2. Total RNA Extraction, Poly(A) Selection and First-Strand cDNA Synthesis
4.3. PCR Amplification of RAS mRNA Transcripts
4.4. Nanopore Sequencing
4.5. Validation of Nanopore Findings with Next-Generation Sequencing
4.6. Post Processing and Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Abankwa, D.; Gorfe, A.A.; Inder, K.; Hancock, J.F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 1130–1135. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Milburn, M.V.; Tong, L.; de Vos, A.M.; Brünger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colicelli, J. Human ras superfamily proteins and related gtpases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The gtpase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Valencia, A.; Chardin, P.; Wittinghofer, A.; Sander, C. The ras protein family: Evolutionary tree and role of conserved amino acids. Biochemistry 1991, 30, 4637–4648. [Google Scholar] [CrossRef]
- Gorfe, A.A.; Grant, B.J.; McCammon, J.A. Mapping the nucleotide and isoform-dependent structural and dynamical features of ras proteins. Structure 2008, 16, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Buhrman, G.; O’Connor, C.; Zerbe, B.; Kearney, B.M.; Napoleon, R.; Kovrigina, E.A.; Vajda, S.; Kozakov, D.; Kovrigin, E.L.; Mattos, C. Analysis of binding site hot spots on the surface of ras gtpase. J. Mol. Biol. 2011, 413, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.M.; Chen-Hwang, M.C.; Hwang, Y.W. Switching nucleotide specificity of ha-ras p21 by a single amino acid substitution at aspartate 119. J. Biol. Chem. 1995, 270, 10002–10007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.A.; Mattos, C. The ras-membrane interface: Isoform-specific differences in the catalytic domain. Mol. Cancer Res. MCR 2015, 13, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.W.; Reid, D.; Parker, J.A.; Salter, S.; Knihtila, R.; Kuzmic, P.; Mattos, C. The small gtpases k-ras, n-ras, and h-ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem. 2017, 292, 12981–12993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Spoerner, M.; Hozsa, C.; Poetzl, J.A.; Reiss, K.; Ganser, P.; Geyer, M.; Kalbitzer, H.R. Conformational states of human rat sarcoma (ras) protein complexed with its natural ligand gtp and their role for effector interaction and gtp hydrolysis. J. Biol. Chem. 2010, 285, 39768–39778. [Google Scholar] [CrossRef] [Green Version]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational modifications of ras proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. Ras isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.L.; Hall, A. Rho gtpases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef]
- Bernards, A.; Settleman, J. Gap control: Regulating the regulators of small gtpases. Trends Cell Biol. 2004, 14, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. Ras-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, E.; Martin-Zanca, D.; Reddy, E.P.; Pierotti, M.A.; Della Porta, G.; Barbacid, M. Malignant activation of a k-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223, 661–664. [Google Scholar] [CrossRef]
- Nakano, H.; Yamamoto, F.; Neville, C.; Evans, D.; Mizuno, T.; Perucho, M. Isolation of transforming sequences of two human lung carcinomas: Structural and functional analysis of the activated c-k-ras oncogenes. Proc. Natl. Acad. Sci. USA 1984, 81, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Huang, J.; Klampfer, L. K-ras, intestinal homeostasis and colon cancer. Curr. Clin. Pharmacol. 2015, 10, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Okabe, T.; Anraku, Y.; Fujisawa, M.; Urabe, A.; Takaku, F. Activation of the c-k-ras oncogene in a human pancreas carcinoma. Biochem. Biophys. Res. Commun. 1985, 127, 168–174. [Google Scholar] [CrossRef]
- Chen, S.; Li, F.; Xu, D.; Hou, K.; Fang, W.; Li, Y. The function of ras mutation in cancer and advances in its drug research. Curr. Pharm. Des. 2019, 25, 1105–1114. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. Ras proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. Targeting ras signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Haller, A.; Lothaire, P.; Meert, A.P.; Noel, S.; et al. The role of ras oncogene in survival of patients with lung cancer: A systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Shibata, D.; Almoguera, C.; Forrester, K.; Dunitz, J.; Martin, S.E.; Cosgrove, M.M.; Perucho, M.; Arnheim, N. Detection of c-k-ras mutations in fine needle aspirates from human pancreatic adenocarcinomas. Cancer Res. 1990, 50, 1279–1283. [Google Scholar] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Vageli, D.; Kiaris, H.; Delakas, D.; Anezinis, P.; Cranidis, A.; Spandidos, D.A. Transcriptional activation of h-ras, k-ras and n-ras proto-oncogenes in human bladder tumors. Cancer Lett. 1996, 107, 241–247. [Google Scholar] [CrossRef]
- Capon, D.J.; Seeburg, P.H.; McGrath, J.P.; Hayflick, J.S.; Edman, U.; Levinson, A.D.; Goeddel, D.V. Activation of ki-ras2 gene in human colon and lung carcinomas by two different point mutations. Nature 1983, 304, 507–513. [Google Scholar] [CrossRef]
- Plowman, S.J.; Arends, M.J.; Brownstein, D.G.; Luo, F.; Devenney, P.S.; Rose, L.; Ritchie, A.M.; Berry, R.L.; Harrison, D.J.; Hooper, M.L.; et al. The k-ras 4a isoform promotes apoptosis but does not affect either lifespan or spontaneous tumor incidence in aging mice. Exp. Cell Res. 2006, 312, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Ye, H.; Hamoudi, R.; Dong, G.; Zhang, W.; Patek, C.E.; Poulogiannis, G.; Arends, M.J. K-ras exon 4a has a tumour suppressor effect on carcinogen-induced murine colonic adenoma formation. J. Pathol. 2010, 220, 542–550. [Google Scholar] [CrossRef]
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-ras 4a and 4b are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [Google Scholar]
- Adamopoulos, P.G.; Theodoropoulou, M.C.; Scorilas, A. Alternative splicing detection tool-a novel perl algorithm for sensitive detection of splicing events, based on next-generation sequencing data analysis. Ann. Transl. Med. 2018, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct rna sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Couso, J.P.; Patraquim, P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017, 18, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.-B. Alternative orfs and small orfs: Shedding light on the dark proteome. Nucleic Acids Res. 2019, 48, 1029–1042. [Google Scholar] [CrossRef]
- Khitun, A.; Ness, T.J.; Slavoff, S.A. Small open reading frames and cellular stress responses. Mol. Omics 2019, 15, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza, S.; Menschaert, G.; Payre, F. In search of lost small peptides. Annu. Rev. Cell Dev. Biol. 2017, 33, 391–416. [Google Scholar] [CrossRef]
- Herrmann, C. Ras-effector interactions: After one decade. Curr. Opin. Struct. Biol. 2003, 13, 122–129. [Google Scholar] [CrossRef]
- Korbie, D.J.; Mattick, J.S. Touchdown pcr for increased specificity and sensitivity in pcr amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for oxford nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (igv): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplification of RAS Transcripts with Touchdown PCR | Primers | |||
|---|---|---|---|---|
| Direction | Sequence (5′→3′) | Length (nt) | Tm (°C) | |
| KRAS | Forward | GCCATTTCGGACTGGGAGC | 19 | 61.12 |
| Reverse | CTCGAACTAATGTATAGAAGGCATCATC | 28 | 59.76 | |
| NRAS | Forward | GCTGTGGTCCTAAATCTGTCCA | 22 | 60.03 |
| Reverse | CTGTGAGACTGAAGACAGCAAC | 22 | 59.20 | |
| HRAS | Forward | GCAGTCGCGCCTGTGAAC | 18 | 62.08 |
| Reverse | GCACCTCCATGTCCTGAGCTT | 21 | 62.40 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamopoulos, P.G.; Tsiakanikas, P.; Boti, M.A.; Scorilas, A. Targeted Long-Read Sequencing Decodes the Transcriptional Atlas of the Founding RAS Gene Family Members. Int. J. Mol. Sci. 2021, 22, 13298. https://doi.org/10.3390/ijms222413298
Adamopoulos PG, Tsiakanikas P, Boti MA, Scorilas A. Targeted Long-Read Sequencing Decodes the Transcriptional Atlas of the Founding RAS Gene Family Members. International Journal of Molecular Sciences. 2021; 22(24):13298. https://doi.org/10.3390/ijms222413298
Chicago/Turabian StyleAdamopoulos, Panagiotis G., Panagiotis Tsiakanikas, Michaela A. Boti, and Andreas Scorilas. 2021. "Targeted Long-Read Sequencing Decodes the Transcriptional Atlas of the Founding RAS Gene Family Members" International Journal of Molecular Sciences 22, no. 24: 13298. https://doi.org/10.3390/ijms222413298
APA StyleAdamopoulos, P. G., Tsiakanikas, P., Boti, M. A., & Scorilas, A. (2021). Targeted Long-Read Sequencing Decodes the Transcriptional Atlas of the Founding RAS Gene Family Members. International Journal of Molecular Sciences, 22(24), 13298. https://doi.org/10.3390/ijms222413298