
Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors


 ,
, 
Abstract
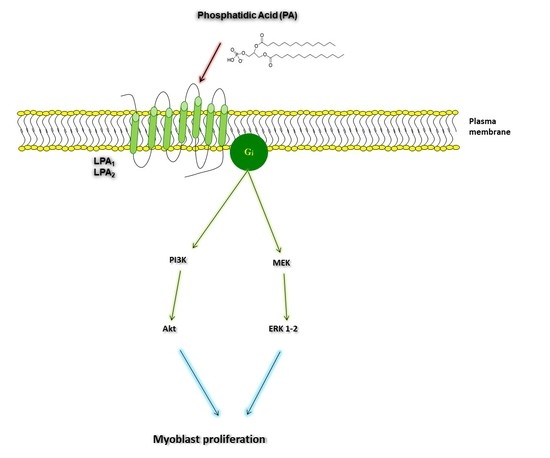
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
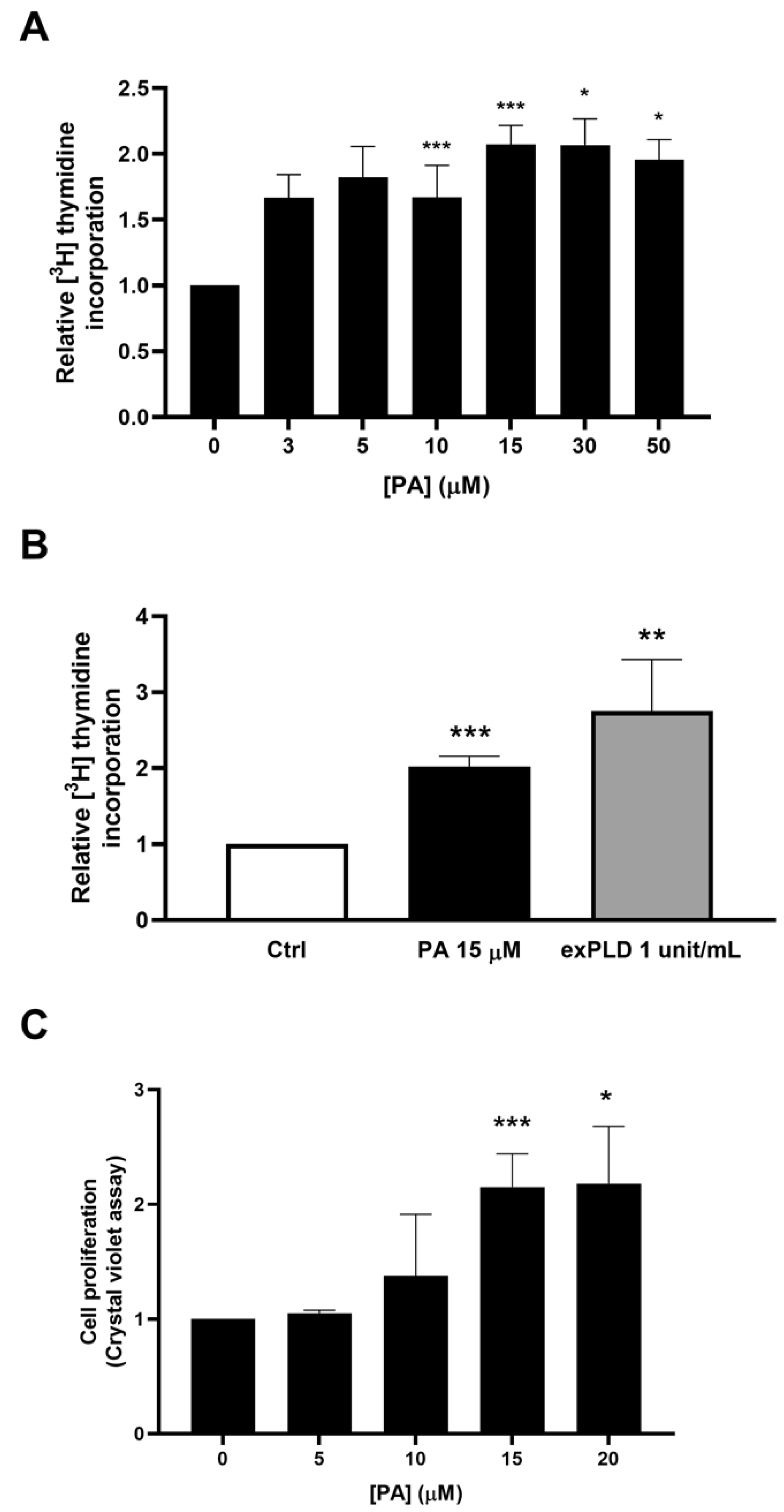
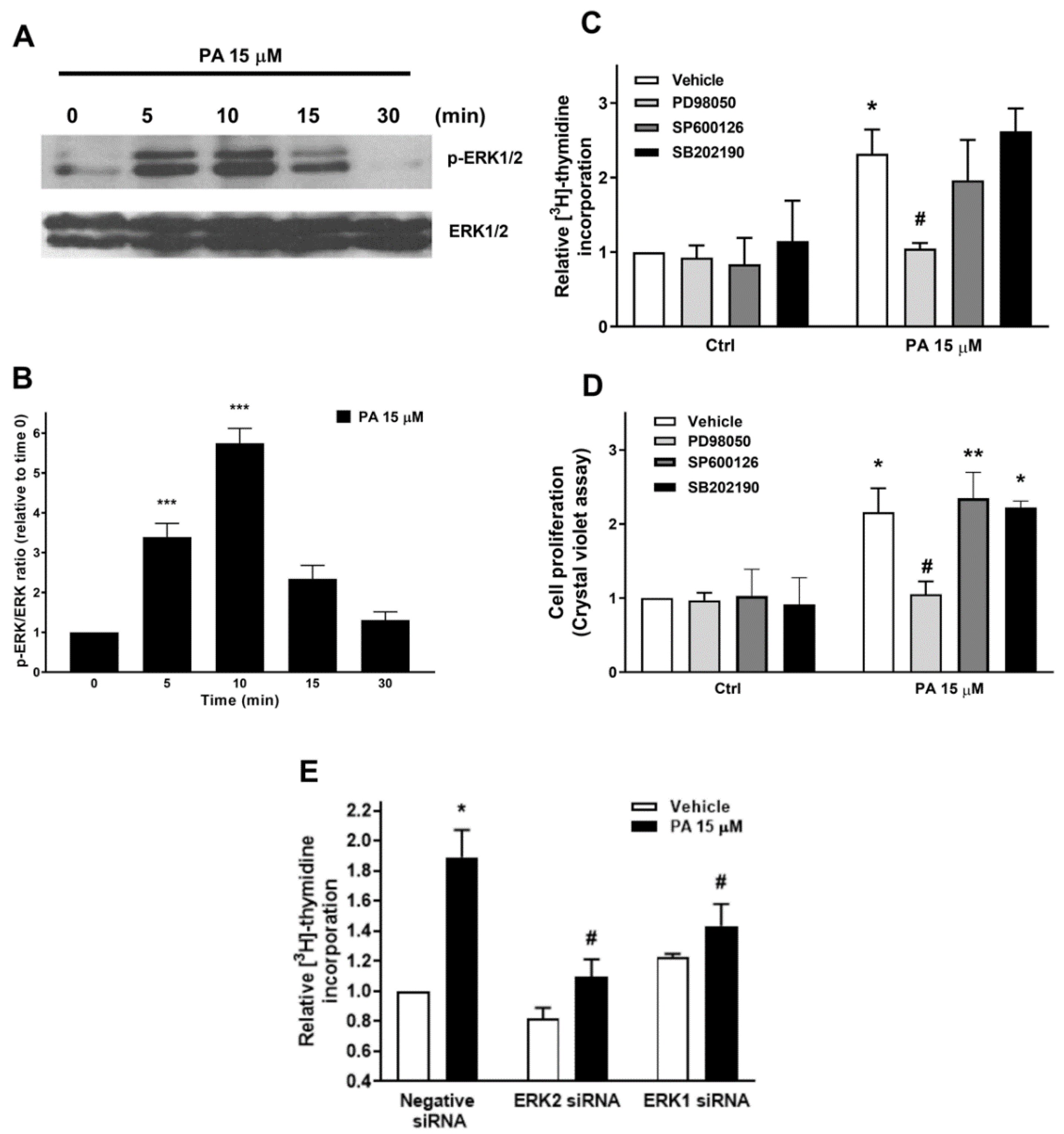
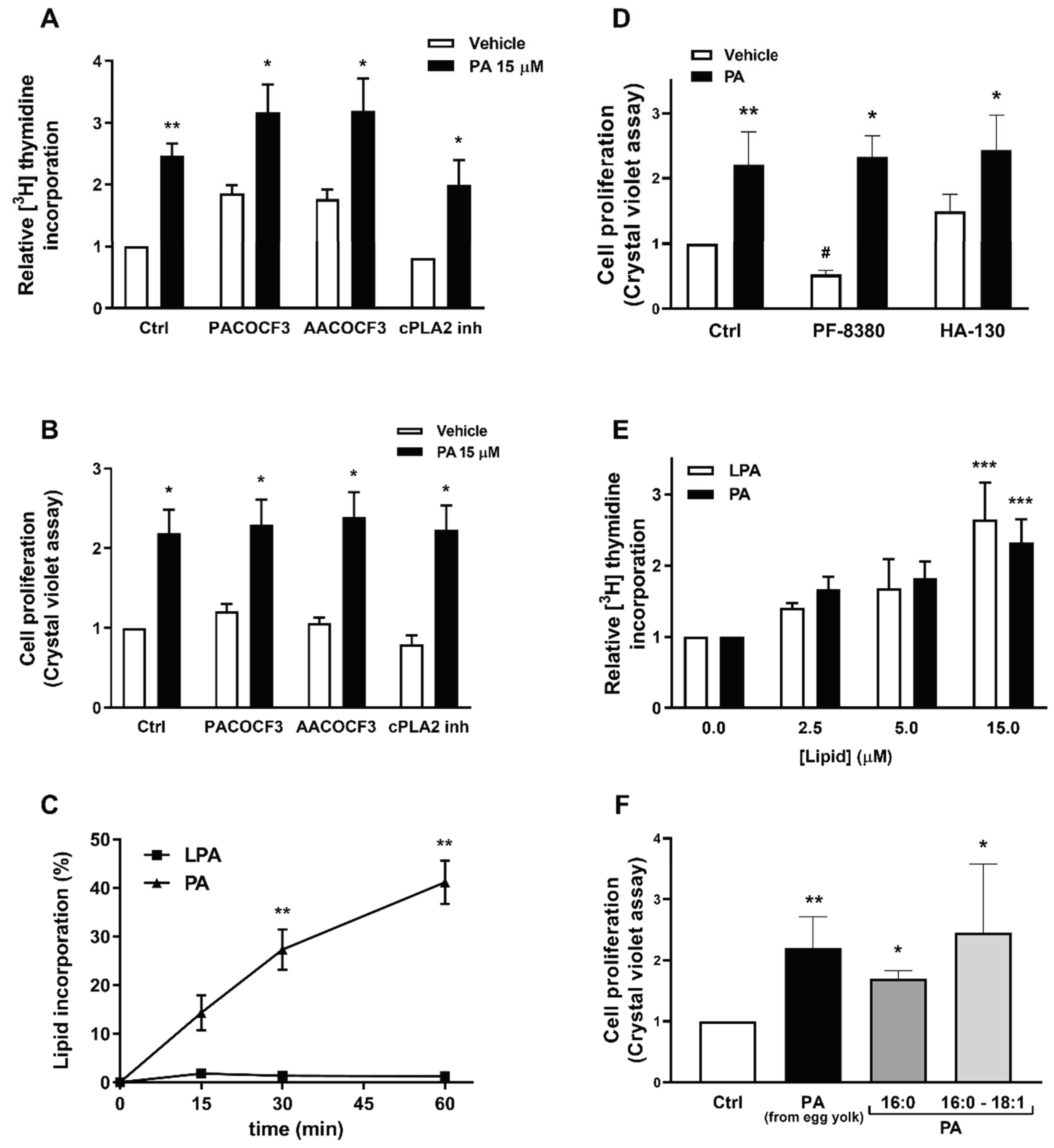
2.1. Phosphatidic Acid Promotes Myoblast DNA Synthesis and Proliferation
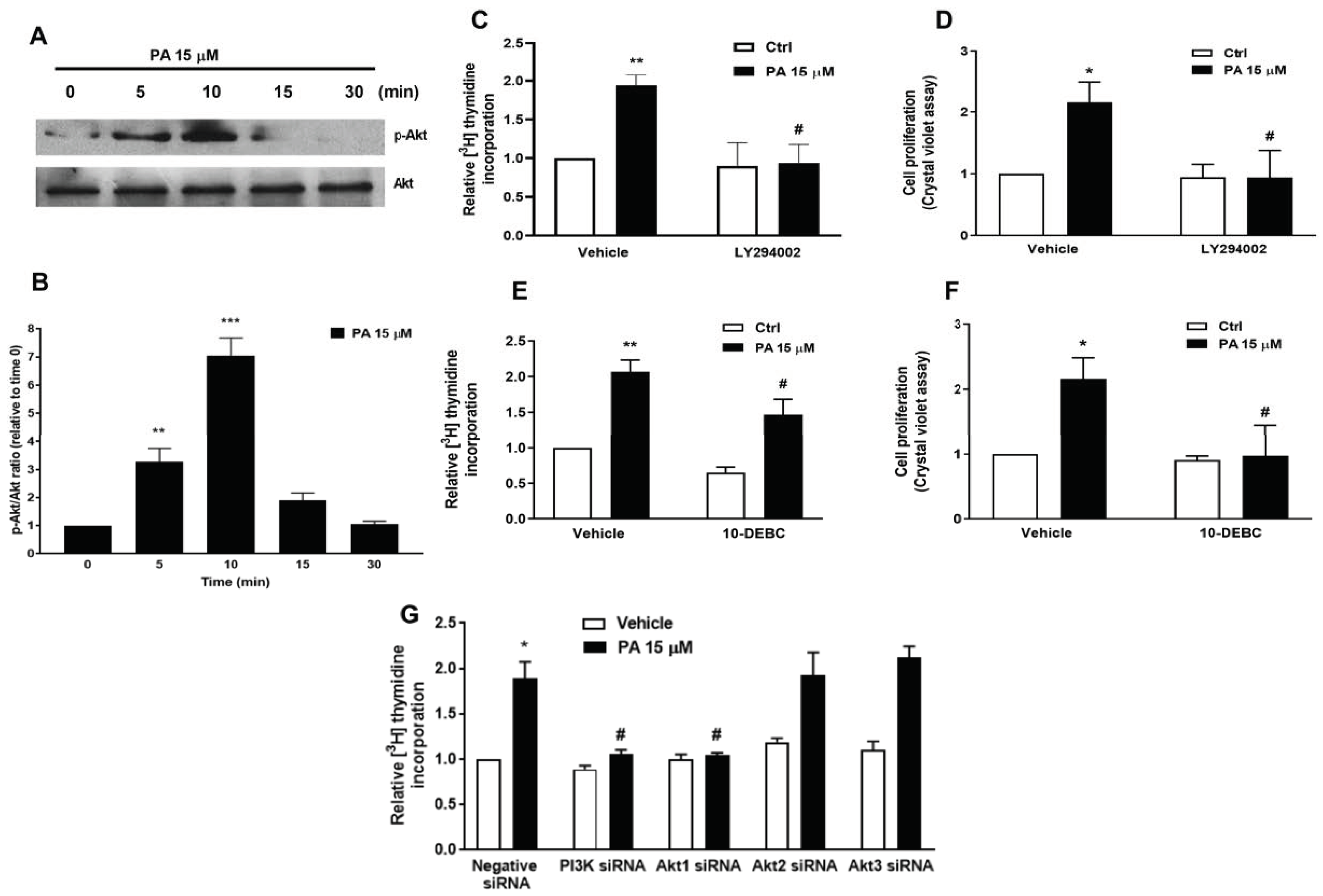
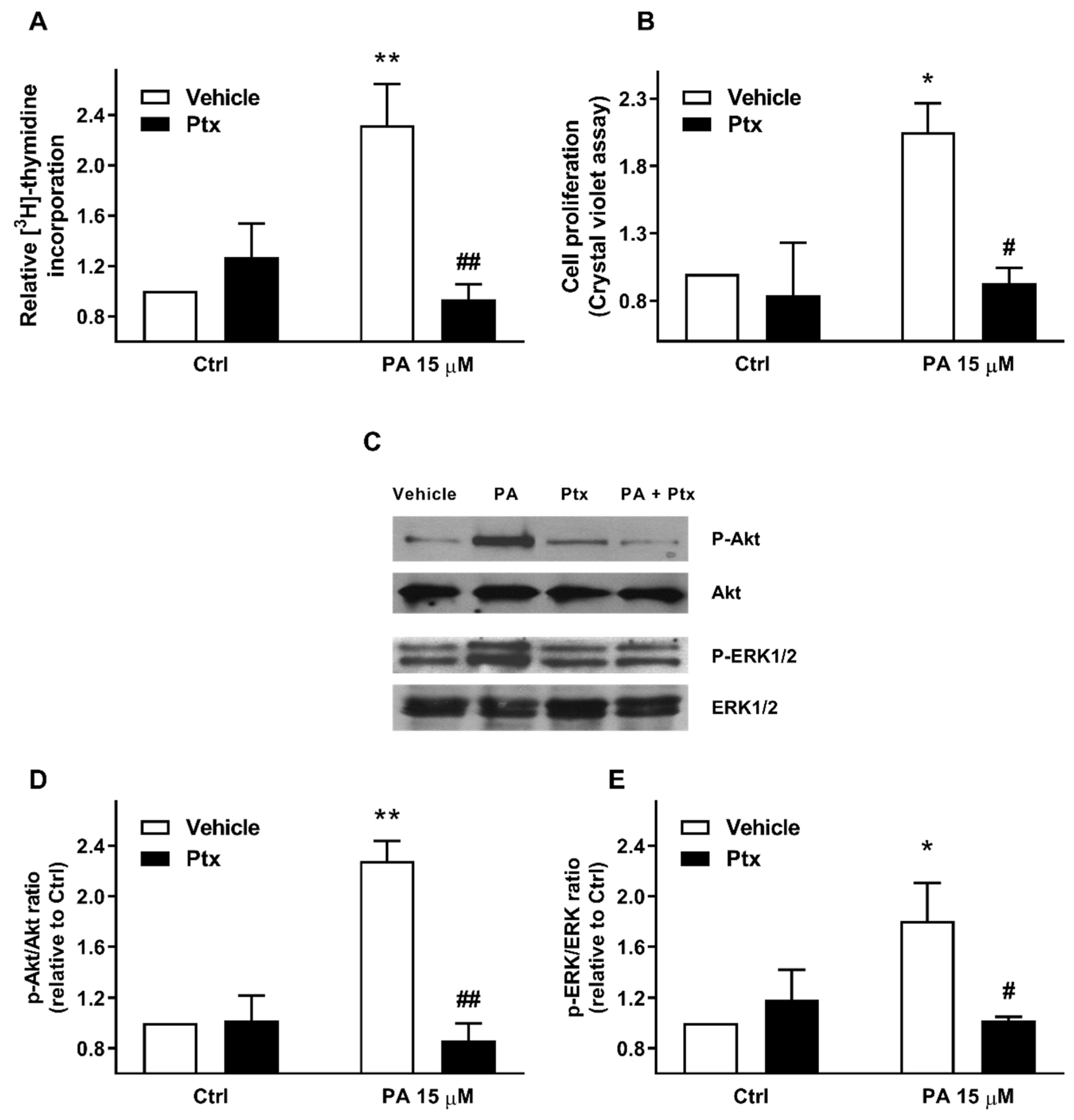
2.2. Phosphatidic Acid Stimulates the PI3-K/Akt and MEK/ERK1-2 Pathways in Myoblasts. Implication in PA-Stimulated Myoblast Proliferation
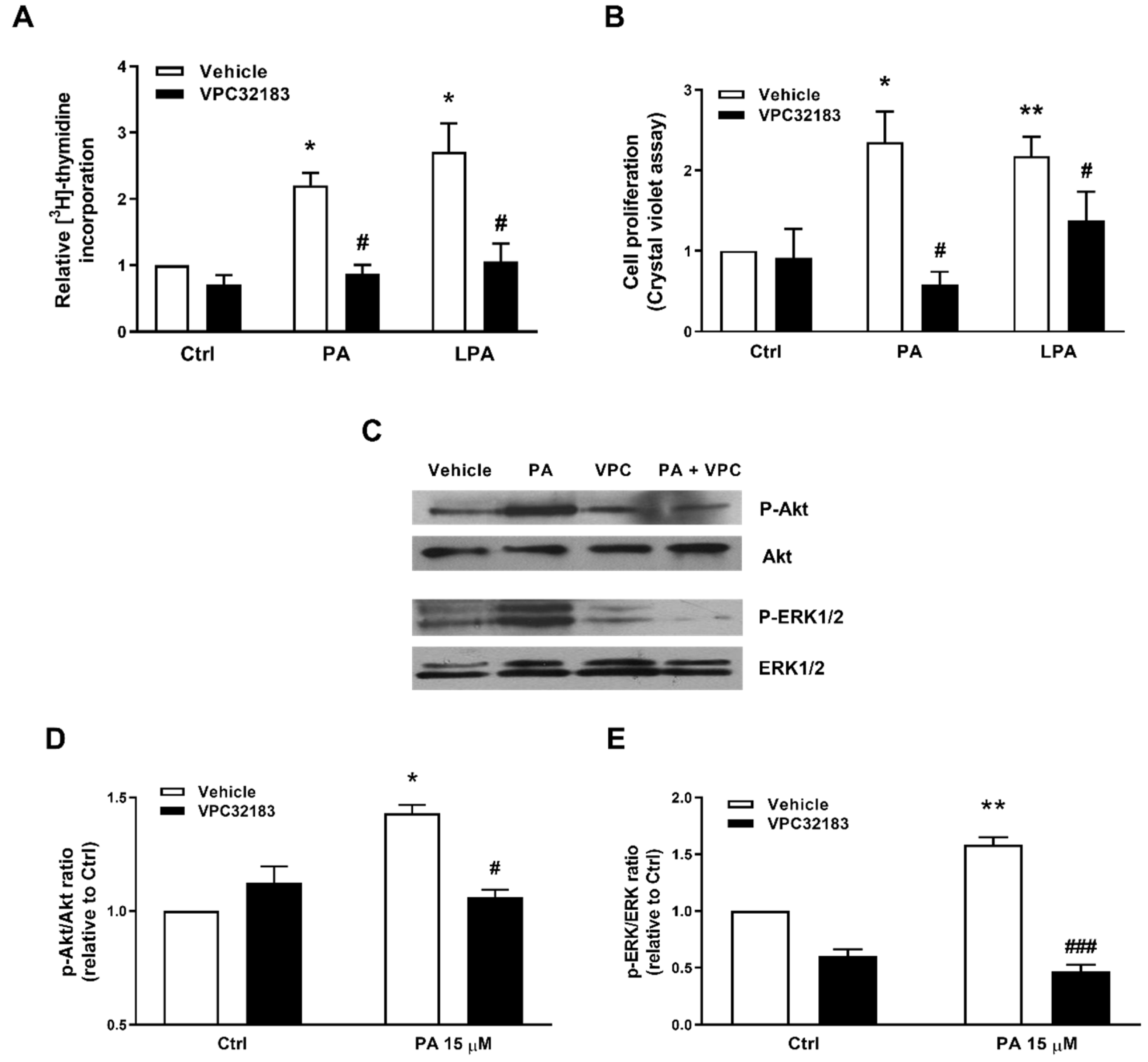
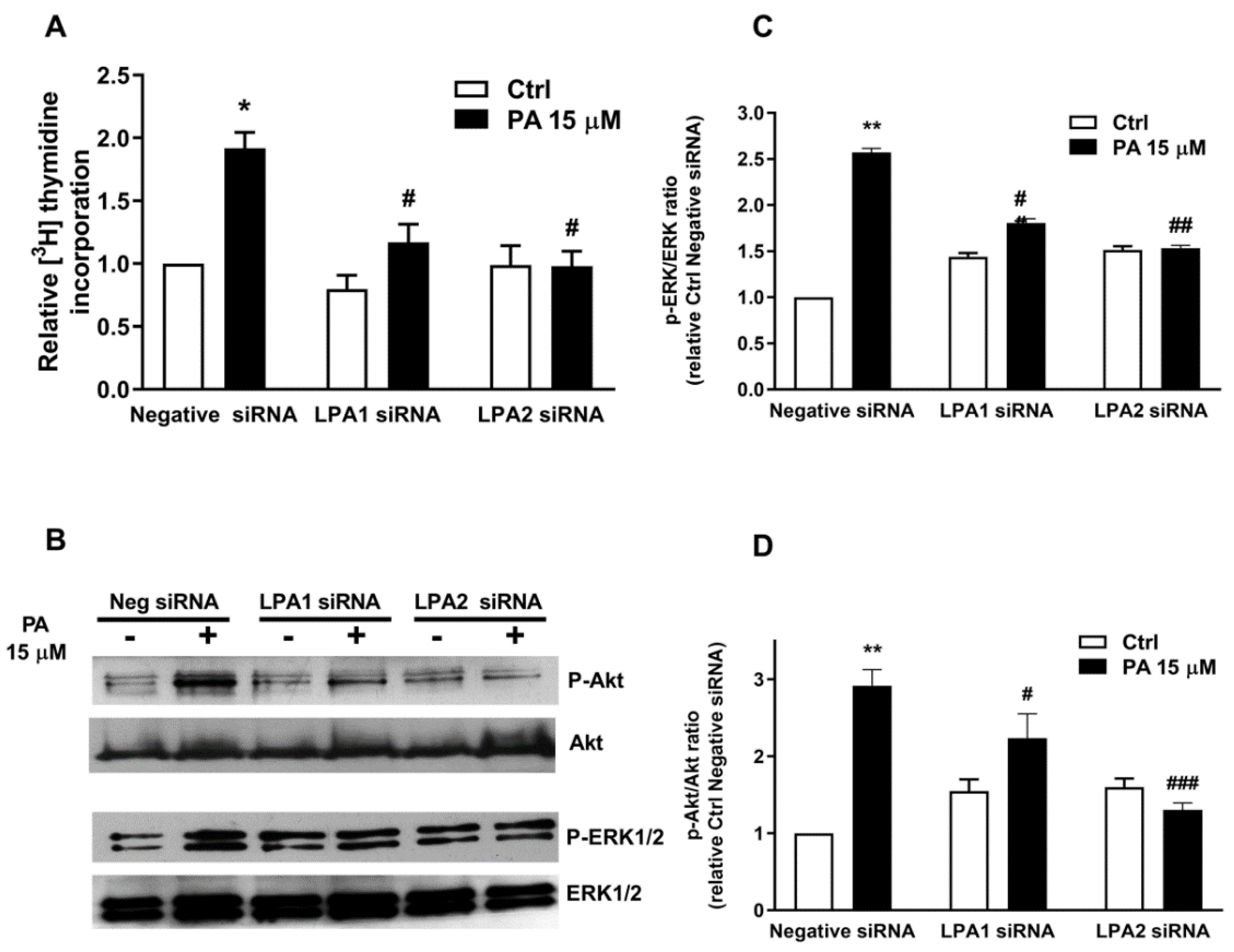
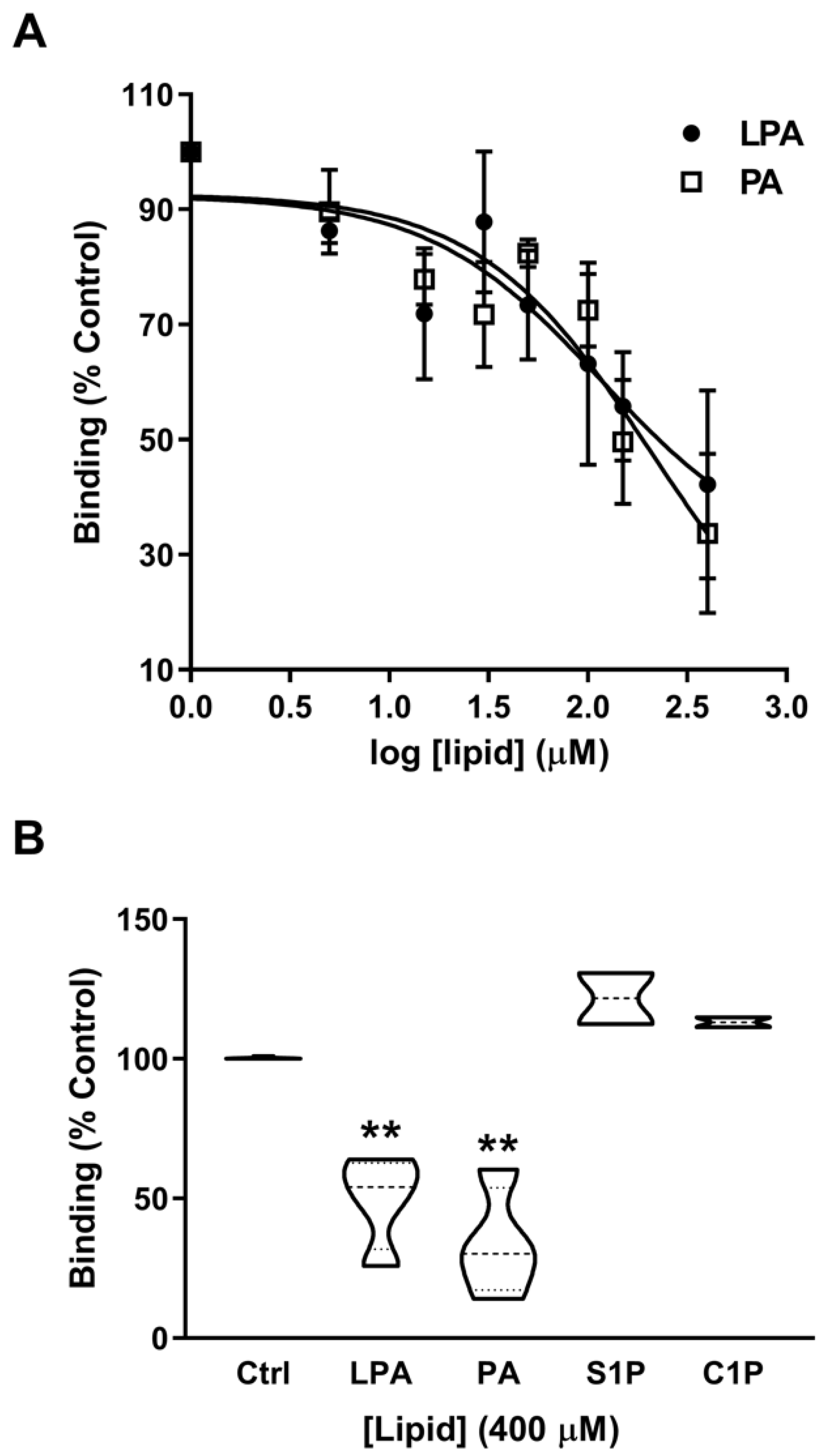
2.3. Phosphatidic Acid Stimulates Myoblast Proliferation Through Interaction with the LPA Receptors LPA1 and LPA2
2.4. PA Stimulates Myoblast Proliferation in the Absence of LPA Formation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Determination of DNA Synthesis
4.4. Determination of Cell Proliferation
4.5. Small Interfering RNA (siRNA)-Mediated Depletion
4.6. Western Blot Analysis
4.7. Phosphatidic Acid Uptake and Metabolism
4.8. Preparation of Cell Membrane and LPA Radioligand-Binding Assay
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brindley, D.N. Intracellular translocation of phosphatidate phosphohydrolase and its possible role in the control of glycerolipid synthesis. Prog. Lipid Res. 1984, 23, 115–133. [Google Scholar] [CrossRef]
- Vance, J.E.; Vance, D.E. Phospholipid biosynthesis in mammalian cells. Biochem. Cell Biol. 2004, 82, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Devaiah, S.P.; Zhang, W.; Welti, R. Signaling functions of phosphatidic acid. Prog. Lipid Res. 2006, 45, 250–278. [Google Scholar] [CrossRef] [PubMed]
- Bargmann, B.O.; Munnik, T. The role of phospholipase D in plant stress responses. Curr. Opin. Plant Biol. 2006, 9, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzolli, R.; Shemon, A.N.; Fang, M.Q.; Hughes, W.E. Phospholipid signalling through phospholipase D and phosphatidic acid. IUBMB Life 2006, 58, 457–461. [Google Scholar] [CrossRef]
- English, D. Phosphatidic acid: A lipid messenger involved in intracellular and extracellular signalling. Cell. Signal. 1996, 8, 341–347. [Google Scholar] [CrossRef]
- English, D.; Cui, Y.; Siddiqui, R.A. Messenger functions of phosphatidic acid. Chem. Phys. Lipids 1996, 80, 117–132. [Google Scholar] [CrossRef]
- Cabot, M.C.; Zhang, Z.C.; Cao, H.T.; Lavie, Y.; Giuliano, A.E.; Van Han, T.; Jones, R.C. Tamoxifen activates cellular phos-pholipase C and D and elicits protein kinase C translocation. Int. J. Cancer. 1997, 70, 567–574. [Google Scholar] [CrossRef]
- Pettitt, T.R.; McDermott, M.; Saqib, K.M.; Shimwell, N.; Wakelam, M.J.O. Phospholipase D1b and D2a generate structurally identical phosphatidic acid species in mammalian cells. Biochem. J. 2001, 360, 707–715. [Google Scholar] [CrossRef]
- Tanguy, E.; Wang, Q.; Moine, H.; Vitale, N. Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Litosch, I.; Pujari, R.; Lee, S.J. Phosphatidic acid regulates signal output by G protein coupled receptors through direct interaction with phospholipase C-β1. Cell. Signal. 2009, 21, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.T.; A Rizzo, M.; Shome, K.; Romero, G. The role of phosphatidic acid in the regulation of the Ras/MEK/Erk signaling cascade. FEBS Lett. 2002, 531, 65–68. [Google Scholar] [CrossRef]
- Winter, J.N.; Fox, T.E.; Kester, M.; Jefferson, L.S.; Kimball, S.R. Phosphatidic acid mediates activation of mTORC1 through the ERK signaling pathway. Am. J. Physiol. Cell Physiol. 2010, 299, C335–C344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornberger, T.A.; Chu, W.K.; Mak, Y.W.; Hsiung, J.W.; Huang, S.A.; Chien, S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 4741–4746. [Google Scholar] [CrossRef] [Green Version]
- Hornberger, T.A.; Sukhija, K.B.; Chien, S. Regulation of mTOR by Mechanically Induced Signaling Events in Skeletal Muscle. Cell Cycle 2006, 5, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Cleland, P.J.; Appleby, G.J.; Rattigan, S.; Clark, M.G. Exercise-induced translocation of protein kinase C and production of diacylglycerol and phosphatidic acid in rat skeletal muscle in vivo. Relationship to changes in glucose transport. J. Biol. Chem. 1989, 264, 17704–17711. [Google Scholar] [CrossRef]
- Shad, B.J.; Smeuninx, B.; Atherton, P.J.; Breen, L. The mechanistic and ergogenic effects of phosphatidic acid in skeletal muscle. Appl. Physiol. Nutr. Metab. 2015, 40, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kassai, A.; Ohmoto, M.; Morito, K.; Kashiwada, Y.; Takaishi, Y.; Urikura, M.; Morishige, J.-I.; Satouchi, K.; Tokumura, A. Quantification of Phosphatidic Acid in Foodstuffs Using a Thin-Layer-Chromatography-Imaging Technique. J. Agric. Food Chem. 2012, 60, 4156–4161. [Google Scholar] [CrossRef]
- Venter, C.; Niesler, C.U. Rapid quantification of cellular proliferation and migration using ImageJ. Biotechniques 2019, 66, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 2004, 15, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression. Cell Cycle 2003, 2, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Cencetti, F.; Bruno, G.; Blescia, S.; Bernacchioni, C.; Bruni, P.; Donati, C. Lysophosphatidic acid stimulates cell migration of satellite cells. A role for the sphingosine kinase/sphingosine 1-phosphate axis. FEBS J. 2014, 281, 4467–4478. [Google Scholar] [CrossRef] [PubMed]
- Jean-Baptiste, G.; Yang, Z.; Khoury, C.; Greenwood, M.T. Lysophosphatidic acid mediates pleiotropic responses in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2005, 335, 1155–1162. [Google Scholar] [CrossRef]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of p2y9/GPR23 as a Novel G Protein-coupled Receptor for Lysophosphatidic Acid, Structurally Distant from the Edg Family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Fujisawa-Sehara, A.; Taki, T.; Arai, K.; Nabeshima, Y. Lysophosphatidic acid and bFGF control different modes in proliferating myoblasts. J. Cell Biol. 1996, 132, 181–193. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Albers, H.M.H.G.; Dong, A.; Van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; Van Tilburg, E.W.; Schuurman, K.; Van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, Y.-C.M.; Krummel, M.F.; Rosen, S.D. Autotaxin through Lysophosphatidic Acid Stimulates Polarization, Motility, and Transendothelial Migration of Naive T Cells. J. Immunol. 2012, 189, 3914–3924. [Google Scholar] [CrossRef] [Green Version]
- Musarò, A. The Basis of Muscle Regeneration. Adv. Biol. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Karalaki, M.; Fili, S.; Philippou, A.; Koutsilieris, M. Muscle regeneration: Cellular and molecular events. In Vivo 2009, 23, 779–796. [Google Scholar] [PubMed]
- Baghdadi, M.B.; Tajbakhsh, S. Regulation and phylogeny of skeletal muscle regeneration. Dev. Biol. 2018, 433, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.; Salloum, D.; Bernfeld, E.; Gorodetsky, E.; Akselrod, A.; Frias, M.A.; Sudderth, J.; Chen, P.-H.; DeBerardinis, R.; Foster, D.A. Lipid sensing by mTOR complexes viade novosynthesis of phosphatidic acid. J. Biol. Chem. 2017, 292, 6303–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-S.; Liu, Y.-W. Mechanical Stretch Induces mTOR Recruitment and Activation at the Phosphatidic Acid-Enriched Macropinosome in Muscle Cell. Front. Cell Dev. Biol. 2019, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, X.; Zhou, H.; Jiang, H.; Mai, K.; He, G. The Mitotic and Metabolic Effects of Phosphatidic Acid in the Primary Muscle Cells of Turbot (Scophthalmus maximus). Front. Endocrinol. 2018, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.S.; Exton, J.H. Activation of actin polymerization by phosphatidic acid derived from phosphatidylcholine in IIC9 fibroblasts. J. Cell Biol. 1993, 123, 1789–1796. [Google Scholar] [CrossRef]
- Pearce, B.; Jakobson, K.; Morrow, C.; Murphy, S. Phosphatidic acid promotes phosphoinositide metabolism and DNA synthesis in cultured cortical astrocytes. Neurochem. Int. 1994, 24, 165–171. [Google Scholar] [CrossRef]
- Ryder, N.S.; Talwar, H.S.; Reynolds, N.J.; Voorhees, J.J.; Fisher, G.J. Phosphatidic acid and phospholipase D both stimulate phosphoinositide turnover in cultured human keratinocytes. Cell. Signal. 1993, 5, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, H.; Sakai, K.; Joho, T.; Izumoto, E.; Fukuda, H. Cell cycle analysis of Chinese hamster ovary cells stimulated by phosphatidic acid in serum-free culture. J. Biosci. Bioeng. 2004, 98, 487–489. [Google Scholar] [CrossRef]
- Zhou, D.; Luini, W.; Bernasconi, S.; Diomede, L.; Salmona, M.; Mantovani, A.; Sozzani, S. Phosphatidic Acid and Lysophosphatidic Acid Induce Haptotactic Migration of Human Monocytes. J. Biol. Chem. 1995, 270, 25549–25556. [Google Scholar] [CrossRef] [Green Version]
- Bond, P. Phosphatidic acid: Biosynthesis, pharmacokinetics, mechanisms of action and effect on strength and body composition in resistance-trained individuals. Nutr. Metab. 2017, 14, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, J.-Y.; Lim, K.-M.; Bae, O.-N.; Chung, S.-M.; Lee, S.-W.; Joo, K.-M.; Lee, S.-D.; Chung, J.-H. Procoagulant and prothrombotic activation of human erythrocytes by phosphatidic acid. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H347–H355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, T.; Schneider, I.; Tölg, R.; Richardt, G. Alpha 1-adrenergic receptor-mediated increase in the mass of phosphatidic acid and 1,2-diacylglycerol in ischemic rat heart. Cardiovasc. Res. 1999, 42, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Kim, Y.-L.; Lee, Y.-K.; Sacket, S.J.; Kim, K.; Kim, H.-L.; Han, M.; Bae, Y.-S.; Okajima, F.; Im, D.-S. Dioleoyl phosphatidic acid increases intracellular Ca2+ through endogenous LPA receptors in C6 glioma and L2071 fibroblasts. Prostaglandins Lipid Mediat. 2007, 83, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Egea-Jimenez, A.L.; Zimmermann, P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J. Lipid Res. 2018, 59, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Kim, Y.-L.; Jo, J.-Y.; Kyeok, K.; Kim, H.-L.; Im, N.-S. Dioeloyl phosphatidic acid induces morphological changes through an endogenous LPA receptor in C6 glioma cells. Arch. Pharmacal Res. 2008, 31, 628–633. [Google Scholar] [CrossRef]
- Frondorf, K.; Henkels, K.M.; Frohman, M.A.; Gomez-Cambronero, J. Phosphatidic Acid Is a Leukocyte Chemoattractant That Acts through S6 Kinase Signaling. J. Biol. Chem. 2010, 285, 15837–15847. [Google Scholar] [CrossRef] [Green Version]
- Bernacchioni, C.; Cencetti, F.; Ouro, A.; Bruno, M.; Gomez-Muñoz, A.; Donati, C.; Bruni, P. Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts. Int. J. Mol. Sci. 2018, 19, 139. [Google Scholar] [CrossRef] [Green Version]
- Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-phosphate stimulates proliferation and migration of satellite cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderton, F.; Sambi, B.; Tate, R.; Pyne, N.J.; Pyne, S. Assessment of agonism at G-protein coupled receptors by phosphatidic acid and lysophosphatidic acid in human embryonic kidney 293 cells. Br. J. Pharmacol. 2001, 134, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Changeux, J.-P.; Christopoulos, A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-S.; Sun, Y.; Arauz, E.; Jiang, Y.; Chen, J. Phosphatidic Acid Activates Mammalian Target of Rapamycin Complex 1 (mTORC1) Kinase by Displacing FK506 Binding Protein 38 (FKBP38) and Exerting an Allosteric Effect. J. Biol. Chem. 2011, 286, 29568–29574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.-S.; Chen, J. PLD regulates myoblast differentiation through the mTOR-IGF2 pathway. J. Cell Sci. 2008, 121, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.-S.; Chen, J. Distinct amino acid–sensing mTOR pathways regulate skeletal myogenesis. Mol. Biol. Cell 2013, 24, 3754–3763. [Google Scholar] [CrossRef]
- Yoon, M.-S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid–sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Wiczer, B.M.; Thomas, G. Phospholipase D and mTORC1: Nutrients Are What Bring Them Together. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Zhu, M.; Zou, J.; Li, T.; O’Brien, S.A.; Zhang, Y.; Ogden, S.; Zhang, W. Differential Roles of Phospholipase D Proteins in FcεRI-Mediated Signaling and Mast Cell Function. J. Immunol. 2015, 195, 4492–4502. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Muñoz, A.; A Duffy, P.; Martin, A.; O’Brien, L.; Byun, H.S.; Bittman, R.; Brindley, D.N. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: Antagonism by cell-permeable ceramides. Mol. Pharmacol. 1995, 47, 833–839. [Google Scholar]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, 343–346. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Myers, D.; Jessup, W.; Cochrane, F.; Byrne, R.; Whitty, G.; Moss, S. Oxidized LDL Can Induce Macrophage Survival, DNA Synthesis, and Enhanced Proliferative Response to CSF-1 and GM-CSF. Arter. Thromb. Vasc. Biol. 1999, 19, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouro, A.; Arana, L.; Rivera, I.-G.; Ordoñez, M.; Gomez-Larrauri, A.; Presa, N.; Simón, J.; Trueba, M.; Gangoiti, P.; Bittman, R.; et al. Phosphatidic acid inhibits ceramide 1-phosphate-stimulated macrophage migration. Biochem. Pharmacol. 2014, 92, 642–650. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Larrauri, A.; Gangoiti, P.; Presa, N.; Dominguez-Herrera, A.; Donati, C.; Bruni, P.; Trueba, M.; Gomez-Muñoz, A.; Ouro, A. Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors. Int. J. Mol. Sci. 2021, 22, 1452. https://doi.org/10.3390/ijms22031452
Gomez-Larrauri A, Gangoiti P, Presa N, Dominguez-Herrera A, Donati C, Bruni P, Trueba M, Gomez-Muñoz A, Ouro A. Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors. International Journal of Molecular Sciences. 2021; 22(3):1452. https://doi.org/10.3390/ijms22031452
Chicago/Turabian StyleGomez-Larrauri, Ana, Patricia Gangoiti, Natalia Presa, Asier Dominguez-Herrera, Chiara Donati, Paola Bruni, Miguel Trueba, Antonio Gomez-Muñoz, and Alberto Ouro. 2021. "Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors" International Journal of Molecular Sciences 22, no. 3: 1452. https://doi.org/10.3390/ijms22031452
APA StyleGomez-Larrauri, A., Gangoiti, P., Presa, N., Dominguez-Herrera, A., Donati, C., Bruni, P., Trueba, M., Gomez-Muñoz, A., & Ouro, A. (2021). Phosphatidic Acid Stimulates Myoblast Proliferation through Interaction with LPA1 and LPA2 Receptors. International Journal of Molecular Sciences, 22(3), 1452. https://doi.org/10.3390/ijms22031452