Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
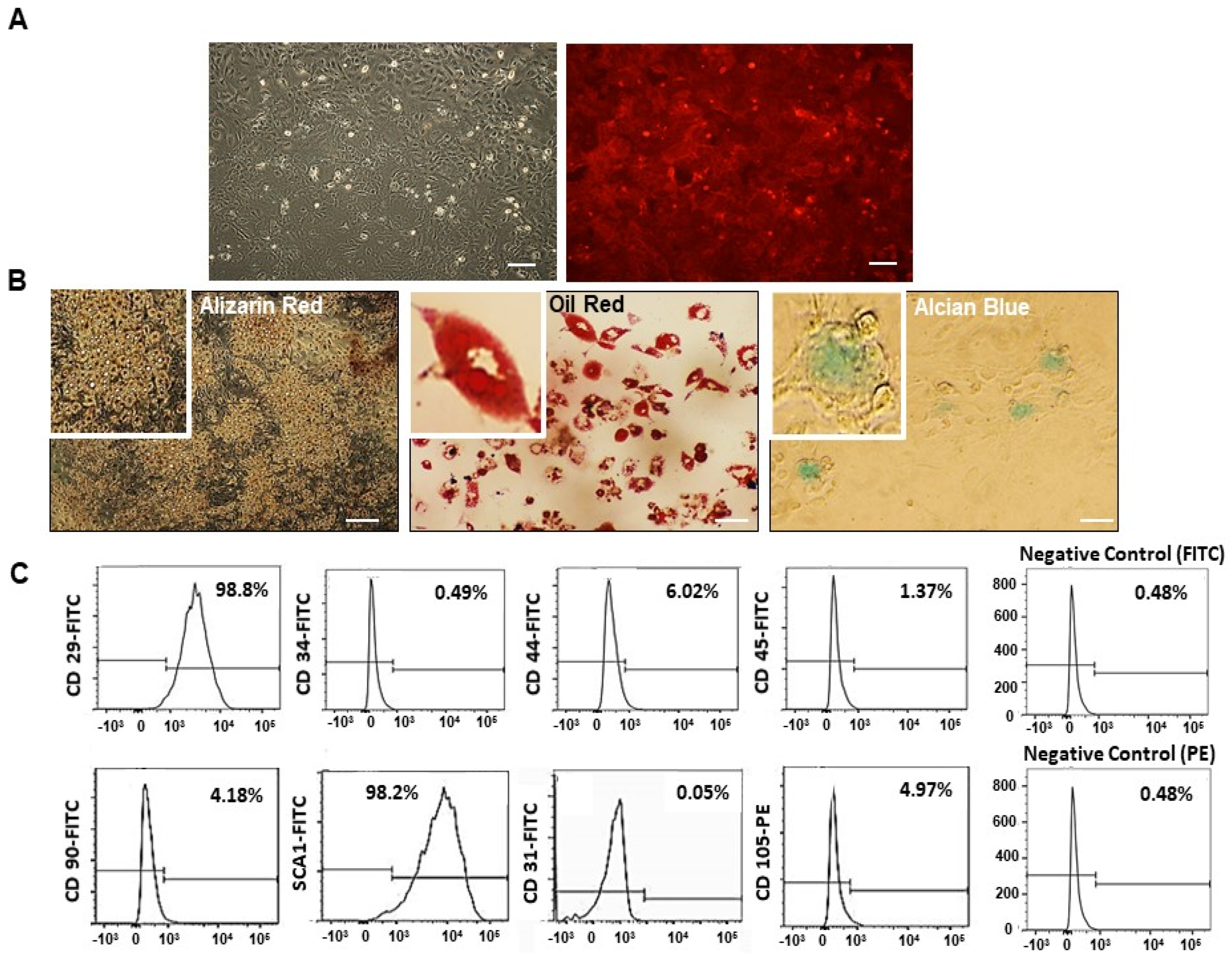
2.1. Mesenchymal Stem Cell (MSC) Characterization
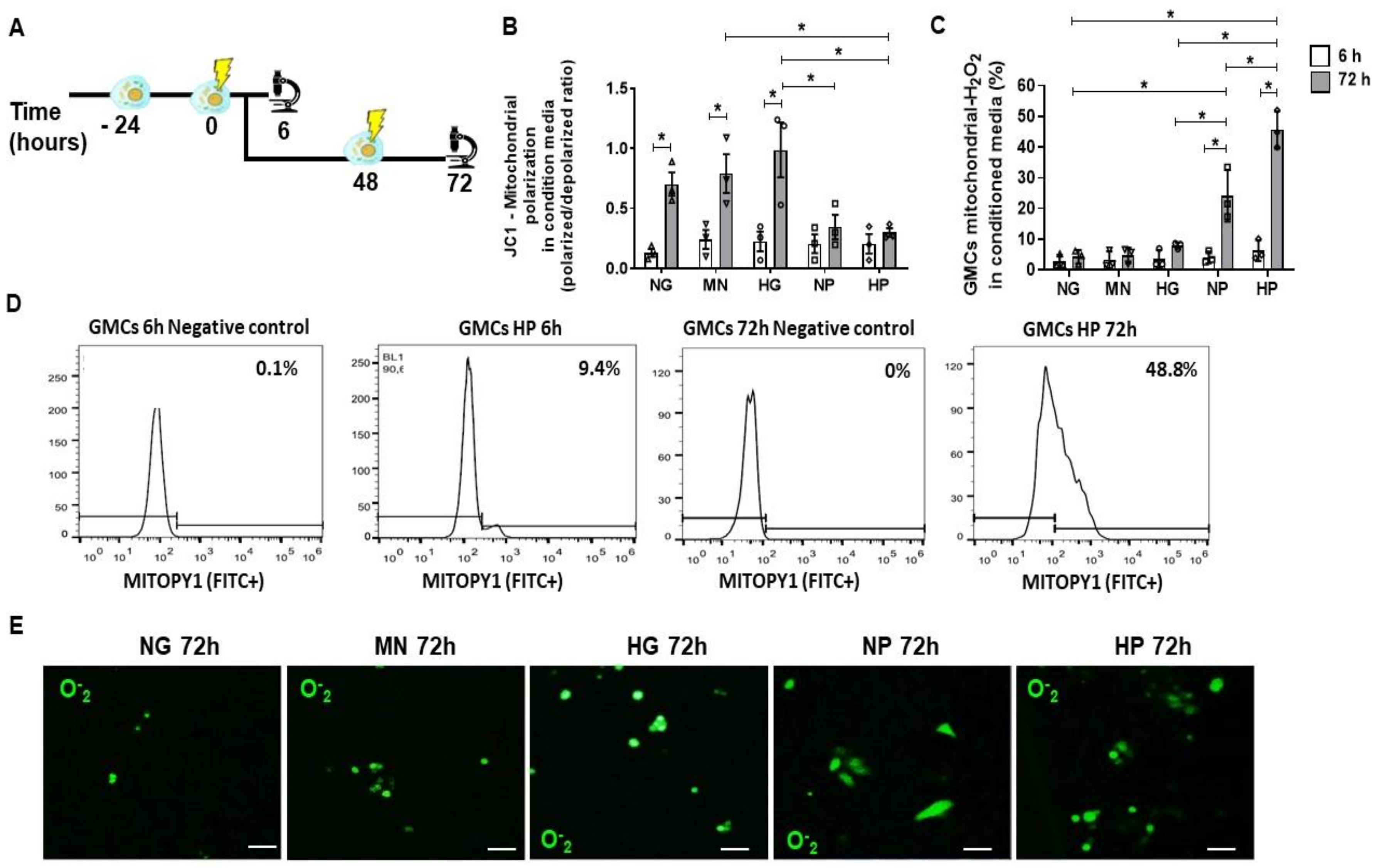
2.2. Mitochondrial Accumulation of H2O2 in Glomerular Mesangial Cells (GMCs) Following Stress Media Conditioning
2.3. Anti-Oxidative Effect of MSCs Co-Culture on GMCs Conditioned with Stress Media
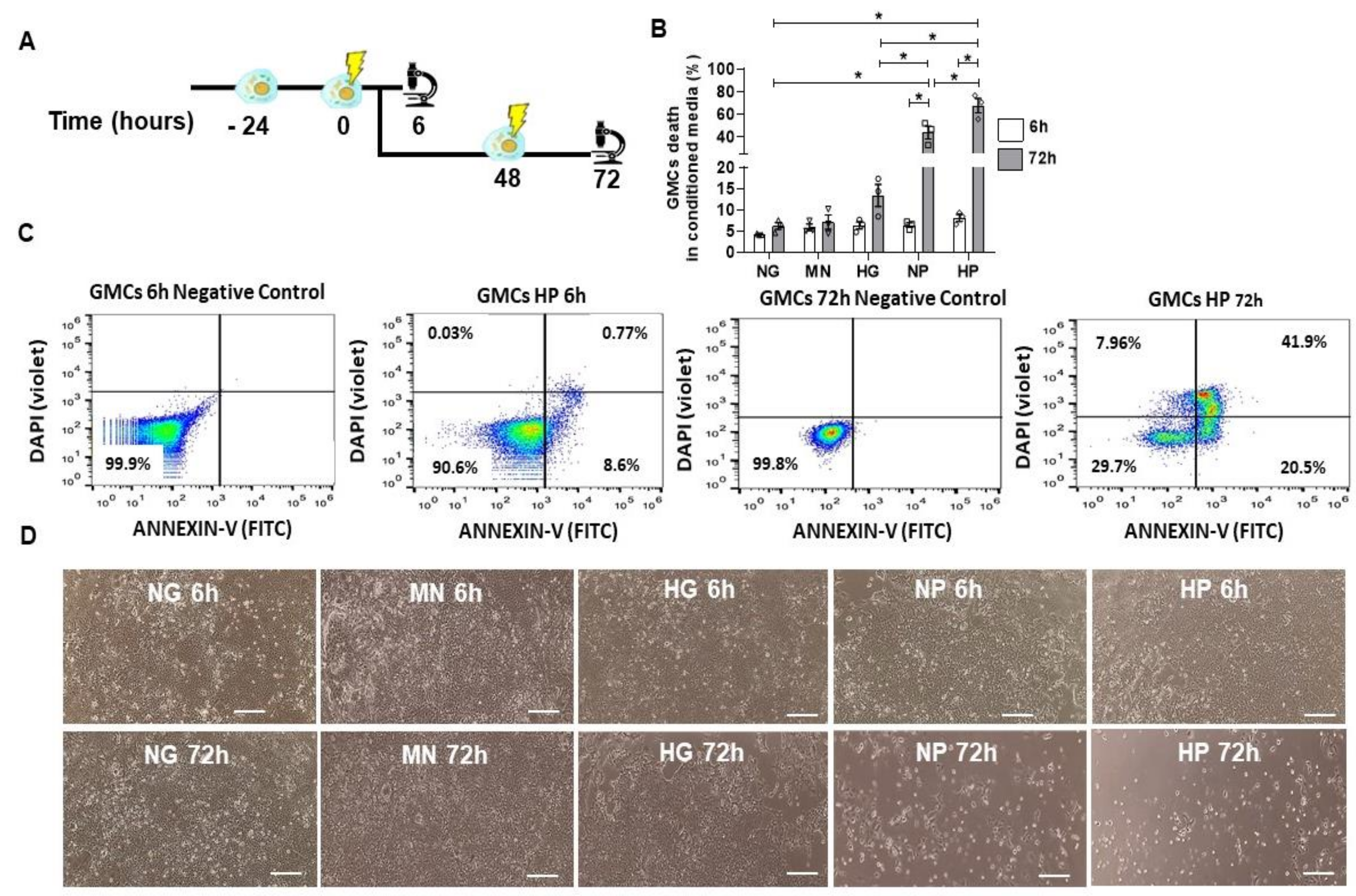
2.4. GMCs Death Induction after Stress-Conditioning Media
2.5. Pro-Survival and Protective Effect of MSCs Co-Culture on Stress-Conditioned GMCs
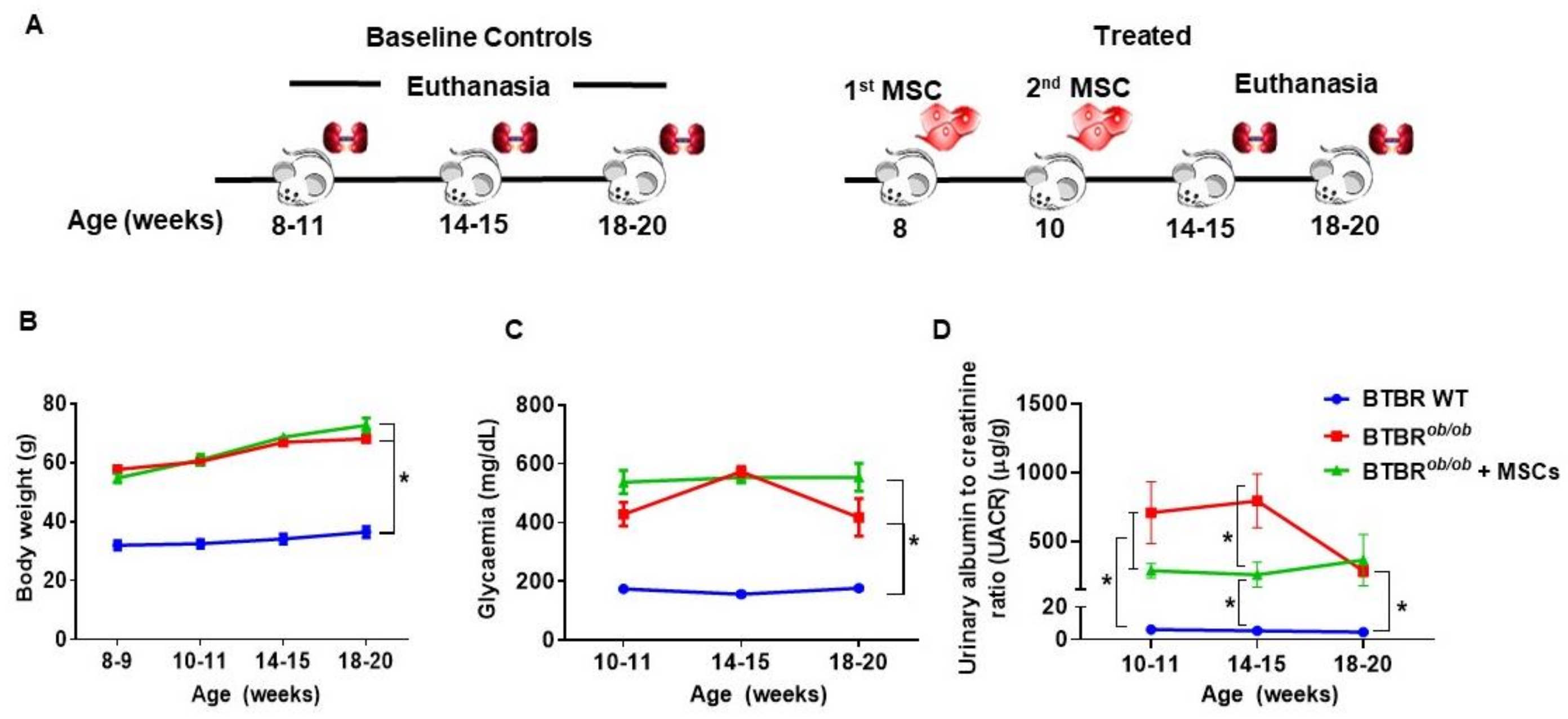
2.6. Metabolic and Renal Functional Parameters in BTBRob/ob Control and MSC-Treated BTBRob/ob Mice
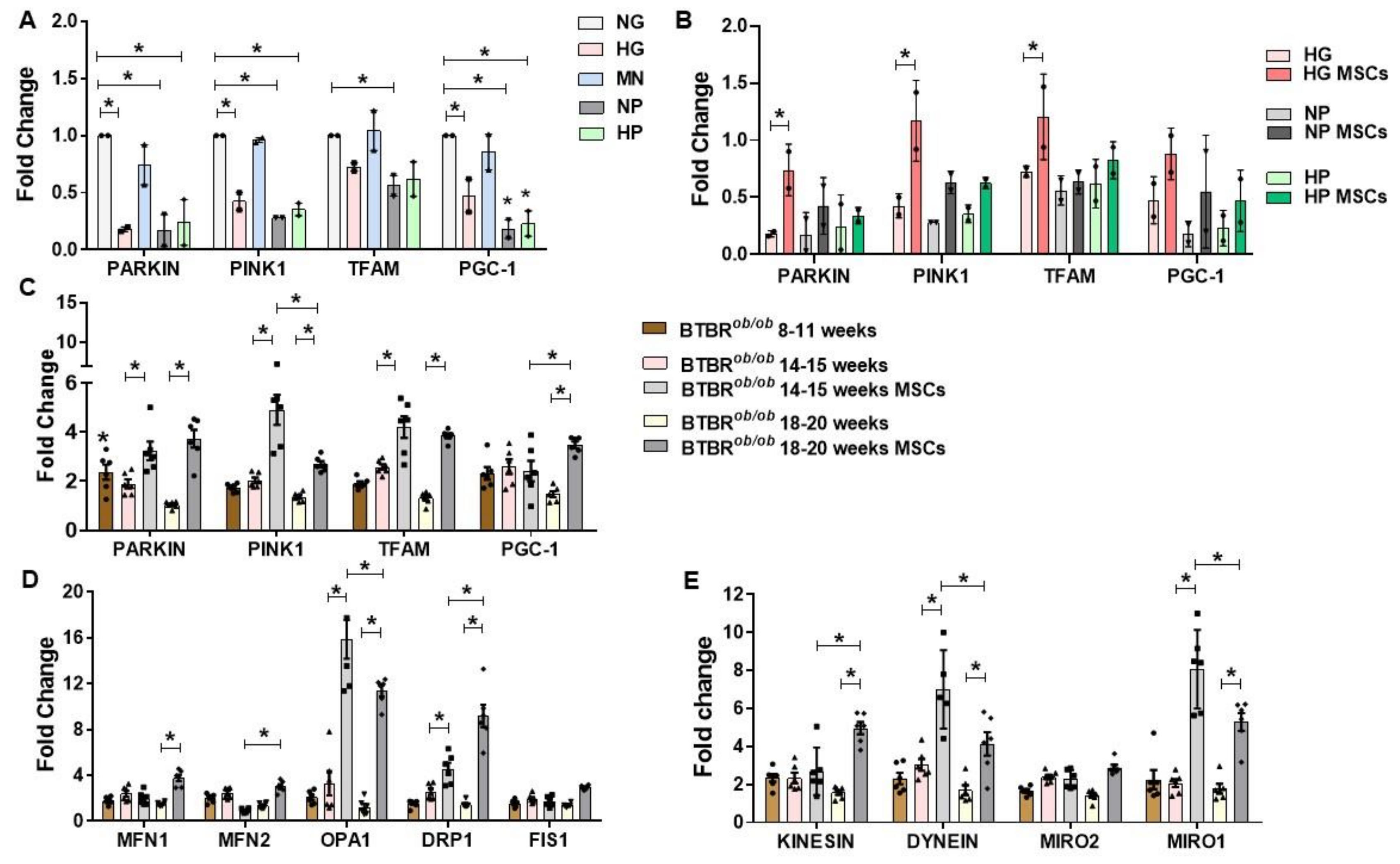
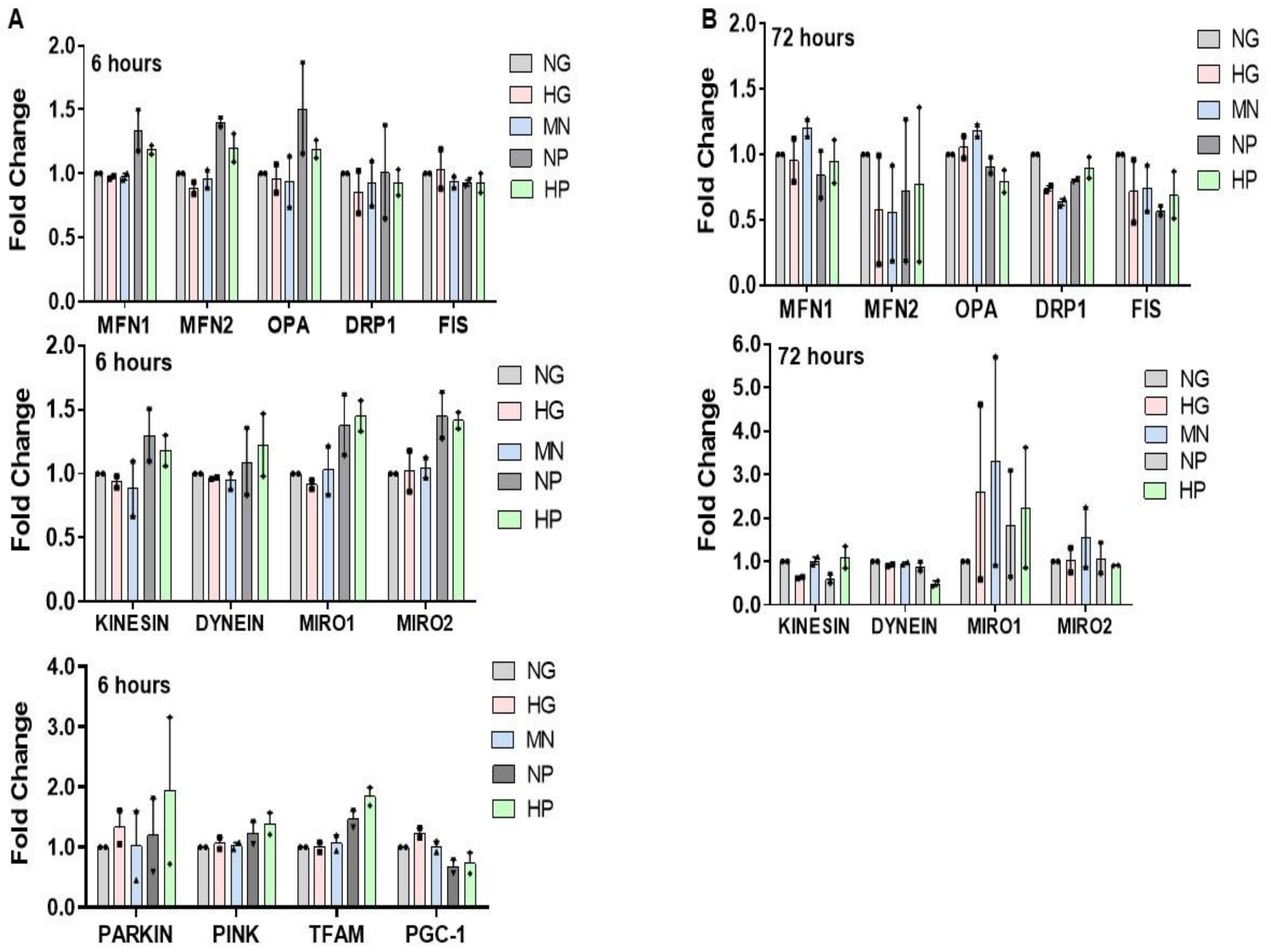
2.7. RNA Expression of Mitochondria Quality Control Program (MQCP)-Related Genes in GMCs and in BTBRob/ob Mice Kidney Cortexes
2.8. MSCs Preserve BTBRob/ob Kidneys from Mesangial Expansion and Podocyte Loss
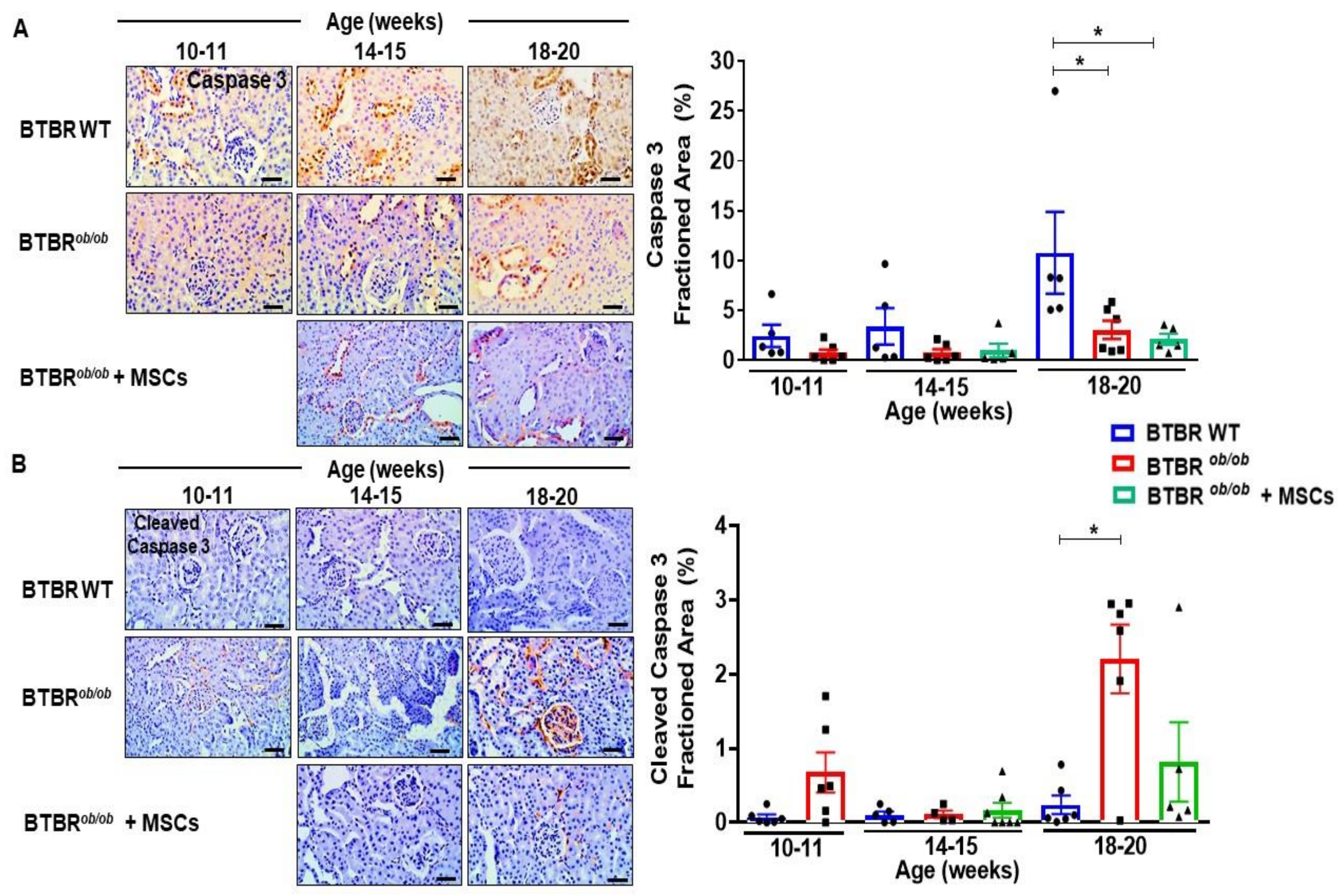
2.9. MSCs Potentially Suppress Caspase-3 Dependent Cell Death over Induction in BTBRob/ob Mice Kidney
2.10. MSCs Therapy Reduces Renal LC3 over Expression and Potentially Diminishes Renal Lipid Peroxidation during DKD Progression
3. Discussion
4. Materials and Methods
4.1. Immortalized Glomerular Mesangial Cells (GMCs)
4.2. Mesenchymal Stem Cells (MSCs)
4.3. BTBRob/ob Mice
4.4. MSCs Administration
4.5. Cell Death
4.6. Mitochondrial-H2O2
4.7. Mitochondrial Membrane Polarization
4.8. Mitochondrial Quality Control Program (MQCP) Targets
4.9. Metabolic and Renal Functional Analysis
4.10. Mesangial Expansion
4.11. Immunohistochemistry (IHC) Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 1–22. [Google Scholar] [CrossRef]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 1–20. [Google Scholar] [CrossRef]
- Anders, H.-J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Gomes, A.P.; Varela, A.T.; Duarte, F.V.; Rolo, A.P.; Palmeira, C.M. Uncovering the beginning of diabetes: The cellular redox status and oxidative stress as starting players in hyperglycemic damage. Mol. Cell. Biochem. 2013, 376, 103–110. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Lindblom, R.; Higgins, G.; Coughlan, M.; de Haan, J.B. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev. Diabet. Stud. Rds 2015, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629. [Google Scholar] [CrossRef]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Bochon, B.; Kozubska, M.; Surygała, G.; Witkowska, A.; Kuźniewicz, R.; Grzeszczak, W.; Wystrychowski, G. Mesenchymal Stem Cells—Potential Applications in Kidney Diseases. Int. J. Mol. Sci. 2019, 20, 2462. [Google Scholar] [CrossRef] [Green Version]
- Peired, A.J.; Sisti, A.; Romagnani, P. Mesenchymal stem cell-based therapy for kidney disease: A review of clinical evidence. Stem Cells Int. 2016, 2016, 4798639. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Cheng, J.; Sun, A.; Li, J.; Wang, W.; Guan, G.; Liu, G.; Su, M. Mesenchymal stem cells transplantation ameliorates glomerular injury in streptozotocin-induced diabetic nephropathy in rats via inhibiting oxidative stress. Diabetes Res. Clin. Pract. 2014, 104, 143–154. [Google Scholar] [CrossRef]
- Lee, S.E.; Jang, J.E.; Kim, H.S.; Jung, M.K.; Ko, M.S.; Kim, M.-O.; Park, H.S.; Oh, W.; Choi, S.J.; Jin, H.J. Mesenchymal stem cells prevent the progression of diabetic nephropathy by improving mitochondrial function in tubular epithelial cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Torres Crigna, A.; Daniele, C.; Gamez, C.; Medina Balbuena, S.; Pastene, D.O.; Nardozi, D.; Brenna, C.; Yard, B.; Gretz, N.; Bieback, K. Stem/stromal cells for treatment of kidney injuries with focus on preclinical models. Front. Med. 2018, 5, 179. [Google Scholar] [CrossRef]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Muhlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2010, 21, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Thomson, S.C.; Vallon, V. Renal Effects of Sodium-Glucose Co-Transporter Inhibitors. Am. J. Med. 2019, 132, S30–S38.e4. [Google Scholar] [CrossRef] [Green Version]
- Rota, C.; Morigi, M.; Imberti, B. Stem Cell Therapies in Kidney Diseases: Progress and Challenges. Int. J. Mol. Sci. 2019, 20, 2790. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Galipeau, J. Manufacturing mesenchymal stromal cells for clinical applications: A survey of Good Manufacturing Practices at US academic centers. Cytotherapy 2019, 21, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Murphy, M.P.; Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Jensen, M.B.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T. Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Yaniv, Y.; Juhaszova, M.; Nuss, H.B.; Wang, S.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Matching ATP supply and demand in mammalian heart: In vivo, in vitro and in silico perspectives. Ann. N. Y. Acad. Sci. 2010, 1188, 133. [Google Scholar] [CrossRef] [Green Version]
- Nagaishi, K.; Mizue, Y.; Chikenji, T.; Otani, M.; Nakano, M.; Konari, N.; Fujimiya, M. Mesenchymal stem cell therapy ameliorates diabetic nephropathy via the paracrine effect of renal trophic factors including exosomes. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Kitamura, S.; Wada, J. Immunomodulatory and regenerative effects of mesenchymal stem cell-derived extracellular vesicles in renal diseases. Int. J. Mol. Sci. 2020, 21, 756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Min, X.-H.; Wang, Q.-Y.; Leung, F.W.; Shi, L.; Zhou, Y.; Yu, T.; Wang, C.-M.; An, G.; Sha, W.-H. Pre-activation of mesenchymal stem cells with TNF-α, IL-1β and nitric oxide enhances its paracrine effects on radiation-induced intestinal injury. Sci. Rep. 2015, 5, 8718. [Google Scholar] [PubMed] [Green Version]
- Mahrouf-Yorgov, M.; Augeul, L.; Da Silva, C.C.; Jourdan, M.; Rigolet, M.; Manin, S.; Ferrera, R.; Ovize, M.; Henry, A.; Guguin, A. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ. 2017, 24, 1224–1238. [Google Scholar] [CrossRef] [Green Version]
- Perico, L.; Morigi, M.; Rota, C.; Breno, M.; Mele, C.; Noris, M.; Introna, M.; Capelli, C.; Longaretti, L.; Rottoli, D. Human mesenchymal stromal cells transplanted into mice stimulate renal tubular cells and enhance mitochondrial function. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Wang, Y.; He, J.; Pei, X.; Zhao, W. Systematic review and meta-analysis of mesenchymal stem/stromal cells therapy for impaired renal function in small animal models. Nephrology 2013, 18, 201–208. [Google Scholar] [CrossRef]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Pulmonary Passage is a Major Obstacle for Intravenous Stem Cell Delivery: The Pulmonary First-Pass Effect. Stem Cells Dev. 2008, 18, 683–692. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Zhao, J.; Bouchentouf, M.; Forner, K.; Birman, E.; Yuan, S.; Boivin, M.-N.; Martineau, D. Human marrow-derived mesenchymal stromal cells decrease cisplatin renotoxicity in vitro and in vivo and enhance survival of mice post-intraperitoneal injection. Am. J. Physiol. Ren. Physiol. 2010, 299, F1288–F1298. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.E.; Capcha, J.M.C.; de Bragança, A.C.; Sanches, T.R.; Gouveia, P.Q.; de Oliveira, P.A.F.; Malheiros, D.M.A.C.; Volpini, R.A.; Santinho, M.A.R.; Santana, B.A.A.; et al. Human umbilical cord-derived mesenchymal stromal cells protect against premature renal senescence resulting from oxidative stress in rats with acute kidney injury. Stem Cell Res. Ther. 2017, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Eleuteri, S.; Fierabracci, A. Insights into the secretome of mesenchymal stem cells and its potential applications. Int. J. Mol. Sci. 2019, 20, 4597. [Google Scholar] [CrossRef] [Green Version]
- Zachar, L.; Bačenková, D.; Rosocha, J. Activation, homing, and role of the mesenchymal stem cells in the inflammatory environment. J. Inflamm. Res. 2016, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar]
- Konari, N.; Nagaishi, K.; Kikuchi, S.; Fujimiya, M. Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.E.; Ezquer, M.E.; Parrau, D.B.; Carpio, D.; Yañez, A.J.; Conget, P.A. Systemic administration of multipotent mesenchymal stromal cells reverts hyperglycemia and prevents nephropathy in type 1 diabetic mice. Biol. Blood Marrow Transplant. 2008, 14, 631–640. [Google Scholar]
- Si, Y.; Zhao, Y.; Hao, H.; Liu, J.; Guo, Y.; Mu, Y.; Shen, J.; Cheng, Y.; Fu, X.; Han, W. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: Identification of a novel role in improving insulin sensitivity. Diabetes 2012, 61, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W.; Löwen, J.; Federico, G.; van den Born, J.; Gröne, E.; Gröne, H.J. Accumulation of worn-out GBM material substantially contributes to mesangial matrix expansion in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2017, 312, F1101–F1111. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, N.; Zhang, L.; Hanyu, Z.; Xueyuan, B.; Fu, B.; Shaoyuan, C.; Zhang, W.; Xuefeng, S.; Li, R. Mesenchymal stem cells protect podocytes from apoptosis induced by high glucose via secretion of epithelial growth factor. Stem Cell Res. Ther. 2013, 4, 103. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Yuan, Y.; Liao, G.; Li, L.; Liu, J.; Chen, Y.; Zhang, J.; Cheng, J.; Lu, Y. Mesenchymal stem cells ameliorate hyperglycemia-induced endothelial injury through modulation of mitophagy. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Yuan, Y.; Shi, M.; Li, L.; Liu, J.; Chen, B.; Chen, Y.; An, X.; Liu, S.; Luo, R.; Long, D. Mesenchymal stem cell-conditioned media ameliorate diabetic endothelial dysfunction by improving mitochondrial bioenergetics via the Sirt1/AMPK/PGC-1α pathway. Clin. Sci. 2016, 130, 2181–2198. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Kornienko, J.S.; Smirnova, I.; Pugovkina, N.; Ivanova, J.S.; Shilina, M.; Grinchuk, T.; Shatrova, A.; Aksenov, N.; Zenin, V.; Nikolsky, N. High doses of synthetic antioxidants induce premature senescence in cultivated mesenchymal stem cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Newell, C.; Sabouny, R.; Hittel, D.; Shutt, T.E.; Khan, A.; Klein, M.S.; Shearer, J. Mesenchymal stem cells shift mitochondrial dynamics and enhance oxidative phosphorylation in recipient cells. Front. Physiol. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lv, J.; Zhang, W.; Li, L.; Lv, J.; Geng, Y.; Yin, A. Combination with miR-124a improves the protective action of BMSCs in rescuing injured rat podocytes from abnormal apoptosis and autophagy. J. Cell. Biochem. 2018, 119, 7166. [Google Scholar] [CrossRef] [PubMed]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.-P.; Hartleben, G. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [Green Version]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.-i.; Haneda, M. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [Green Version]
- Pichaiwong, W.; Hudkins, K.L.; Wietecha, T.; Nguyen, T.Q.; Tachaudomdach, C.; Li, W.; Askari, B.; Kobayashi, T.; O’Brien, K.D.; Pippin, J.W. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1088–1102. [Google Scholar] [CrossRef] [Green Version]
- Grohová, A.; Dáňová, K.; Špíšek, R.; Palová-Jelínková, L. Cell based therapy for type 1 diabetes: Should we take hyperglycemia into account? Front. Immunol. 2019, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Paulini, J.; Higuti, E.; Bastos, R.; Gomes, S.A.; Rangel, E.B. Mesenchymal stem cells as therapeutic candidates for halting the progression of diabetic nephropathy. Stem Cells Int. 2016, 2016, 9521629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sávio-Silva, C.; Beyerstedt, S.; Soinski-Sousa, P.E.; Casaro, E.B.; Balby-Rocha, M.T.A.; Simplício-Filho, A.; Alves-Silva, J.; Rangel, É.B. Mesenchymal Stem Cell Therapy for Diabetic Kidney Disease: A Review of the Studies Using Syngeneic, Autologous, Allogeneic, and Xenogeneic Cells. Stem Cells Int. 2020. [Google Scholar] [CrossRef]
- Cho, J.; D’Antuono, M.; Glicksman, M.; Wang, J.; Jonklaas, J. A review of clinical trials: Mesenchymal stem cell transplant therapy in type 1 and type 2 diabetes mellitus. Am. J. Stem Cells 2018, 7, 82. [Google Scholar] [PubMed]
- Miroshnichenko, S.; Usynin, I.; Dudarev, A.; Nimaev, V.; Solovieva, A. Apolipoprotein AI Supports MSCs Survival under Stress Conditions. Int. J. Mol. Sci. 2020, 21, 4062. [Google Scholar] [CrossRef]
- Ooi, Y.Y.; Rahmat, Z.A.; Jose, S.; Ramasamy, R.; Vidyadaran, S. Immunophenotype and differentiation capacity of bone marrow-derived mesenchymal stem cells from CBA/Ca, ICR and Balb/c mice. World J. Stem Cells 2013, 5, 34–42. [Google Scholar] [CrossRef]
- Rangel, E.B.; Gomes, S.A.; Dulce, R.A.; Premer, C.; Rodrigues, C.O.; Kanashiro-Takeuchi, R.M.; Oskouei, B.; Carvalho, D.A.; Ruiz, P.; Reiser, J.; et al. C-Kit+ Cells Isolated from Developing Kidneys Are a Novel Population of Stem Cells with Regenerative Potential. Stem Cells 2013, 31, 1644–1656. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for defiing multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherpay 2006, 8, 315–317. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sávio-Silva, C.; Soinski-Sousa, P.E.; Simplício-Filho, A.; Bastos, R.M.C.; Beyerstedt, S.; Rangel, É.B. Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity. Int. J. Mol. Sci. 2021, 22, 1546. https://doi.org/10.3390/ijms22041546
Sávio-Silva C, Soinski-Sousa PE, Simplício-Filho A, Bastos RMC, Beyerstedt S, Rangel ÉB. Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity. International Journal of Molecular Sciences. 2021; 22(4):1546. https://doi.org/10.3390/ijms22041546
Chicago/Turabian StyleSávio-Silva, Christian, Poliana E. Soinski-Sousa, Antônio Simplício-Filho, Rosana M. C. Bastos, Stephany Beyerstedt, and Érika Bevilaqua Rangel. 2021. "Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity" International Journal of Molecular Sciences 22, no. 4: 1546. https://doi.org/10.3390/ijms22041546
APA StyleSávio-Silva, C., Soinski-Sousa, P. E., Simplício-Filho, A., Bastos, R. M. C., Beyerstedt, S., & Rangel, É. B. (2021). Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity. International Journal of Molecular Sciences, 22(4), 1546. https://doi.org/10.3390/ijms22041546