
Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Clinical Parameters from Human Explanted Hearts
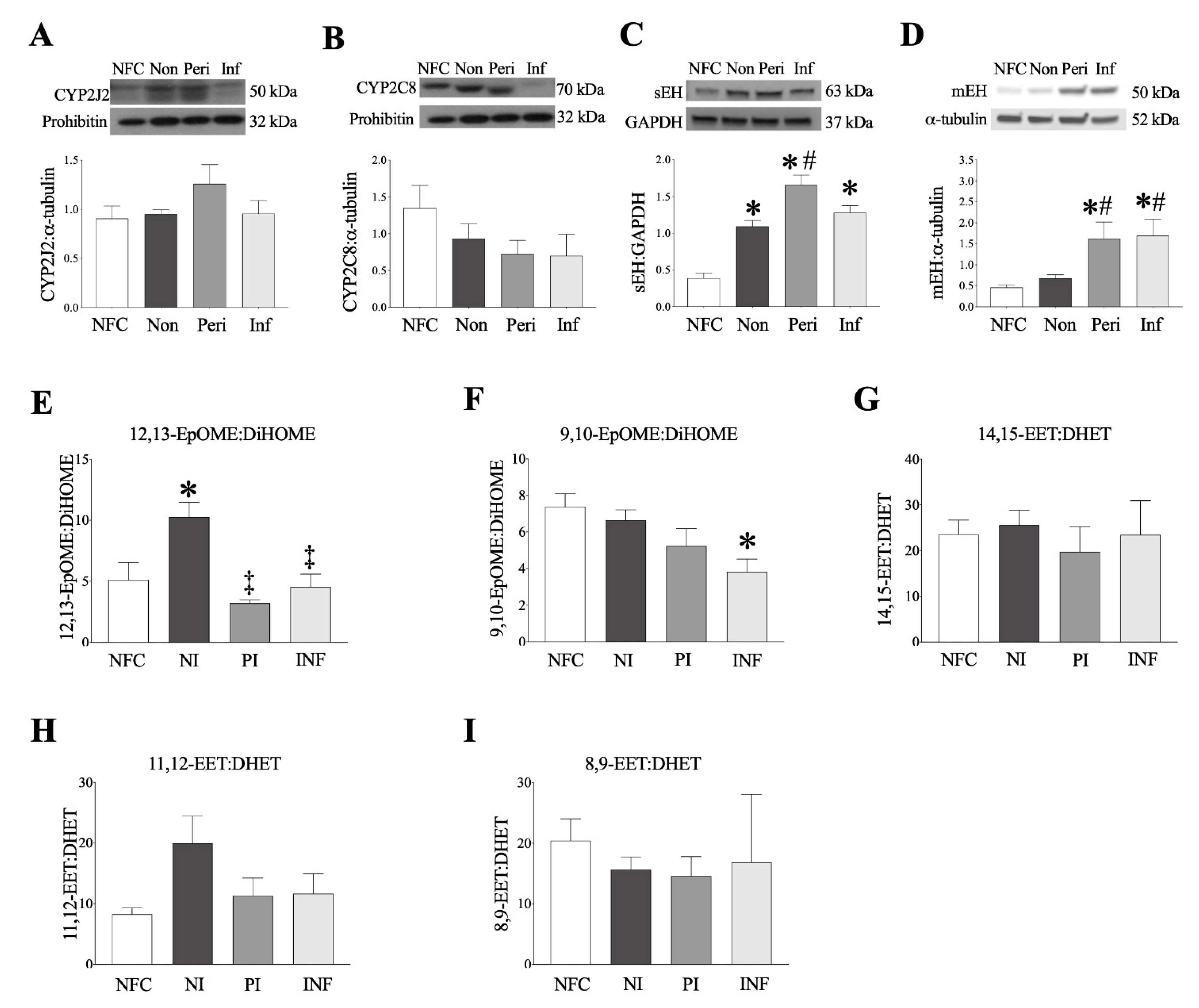
2.2. Epoxide Hydrolases Are Upregulated in Ischemic Human Left Ventricle (LV) Tissues
2.3. Human Left Ventricle Demonstrates Marked Changes in Oxylipid Metabolism Post-MI
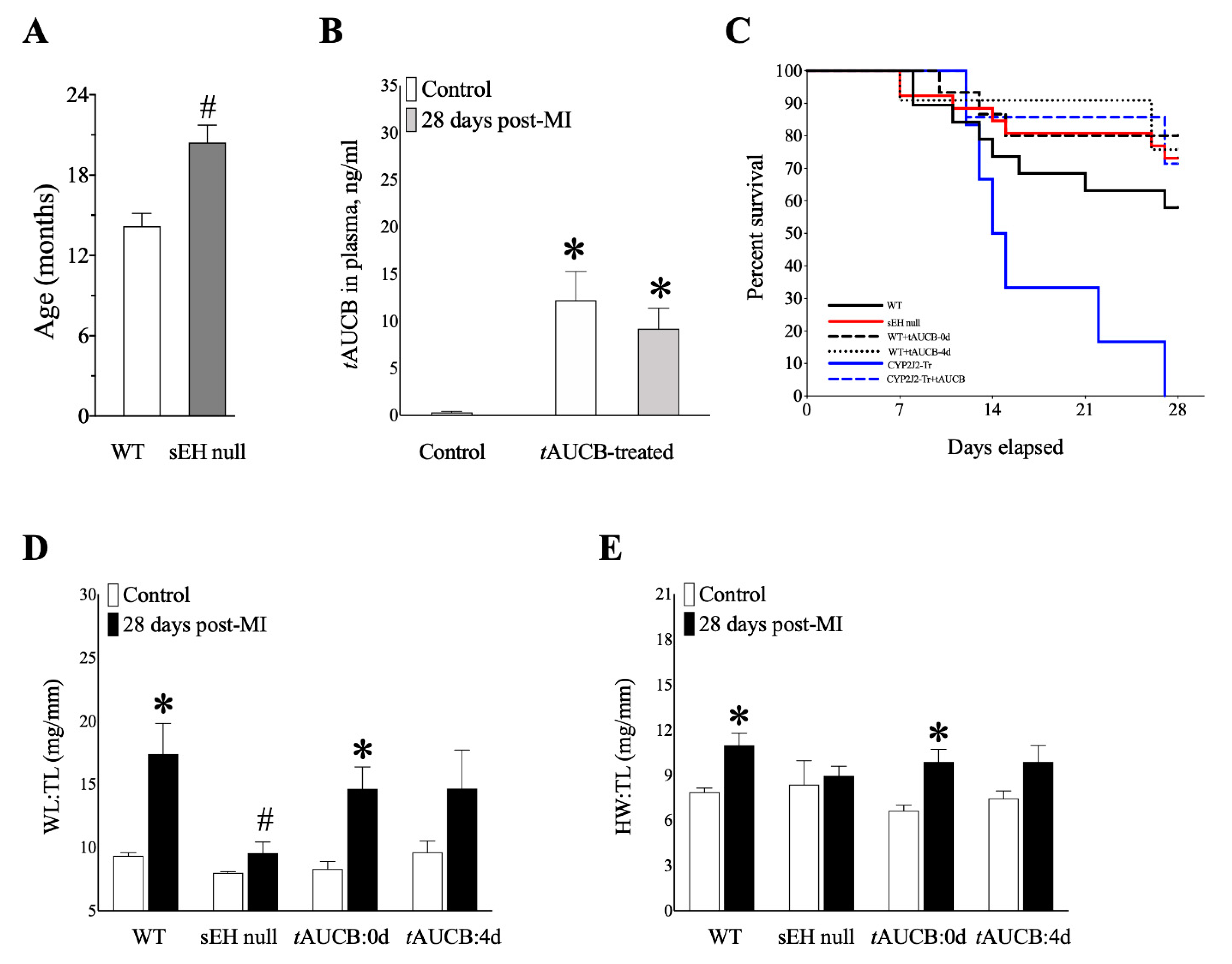
2.4. Genetic Deletion and Pharmacologic Inhibition of sEH Preserves Survival of Mice Post-MI
2.5. sEH Genetic Deletion and tAUCB Pre-Treatment Protects Cardiac Function in Female Mice Post-MI
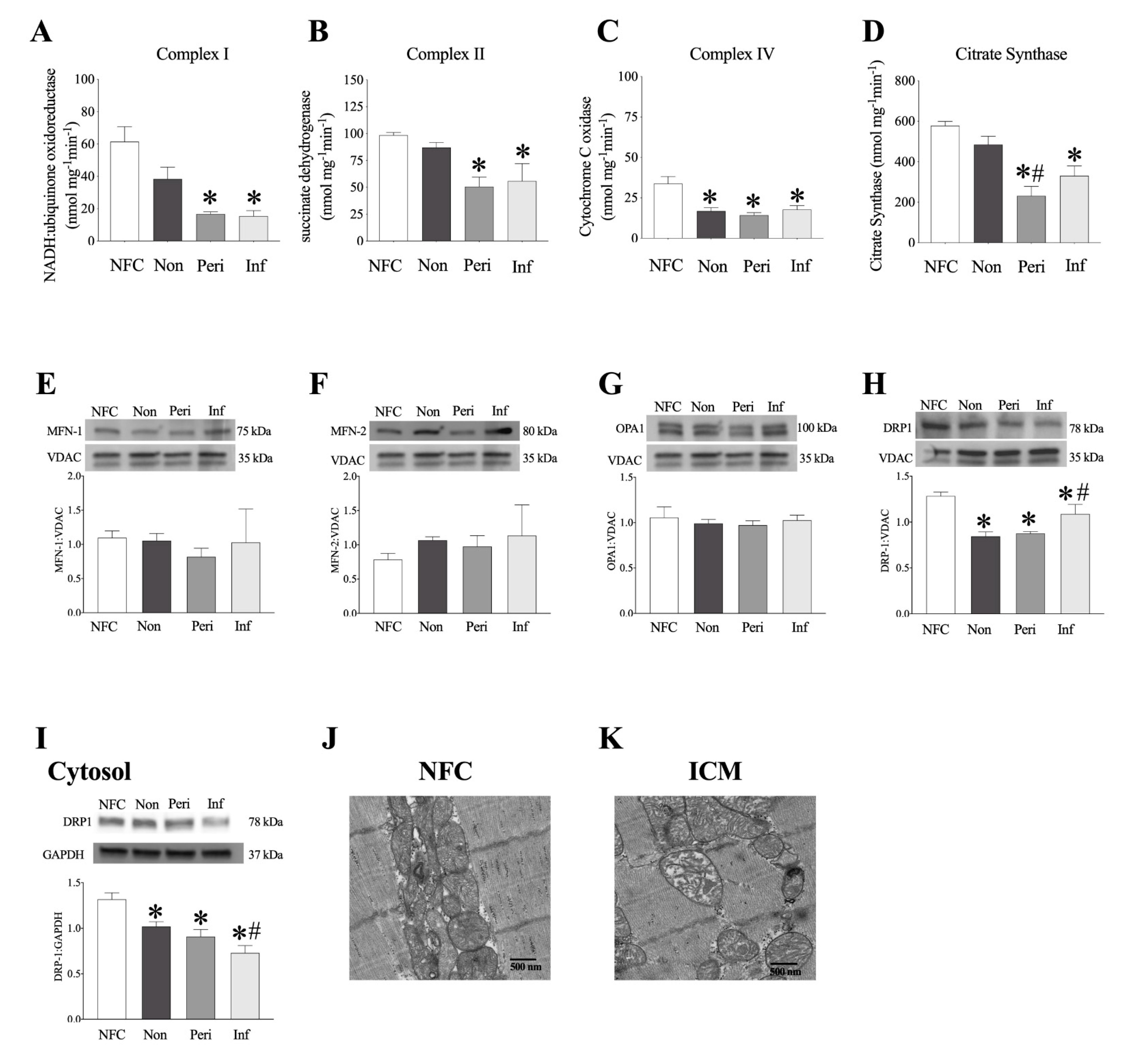
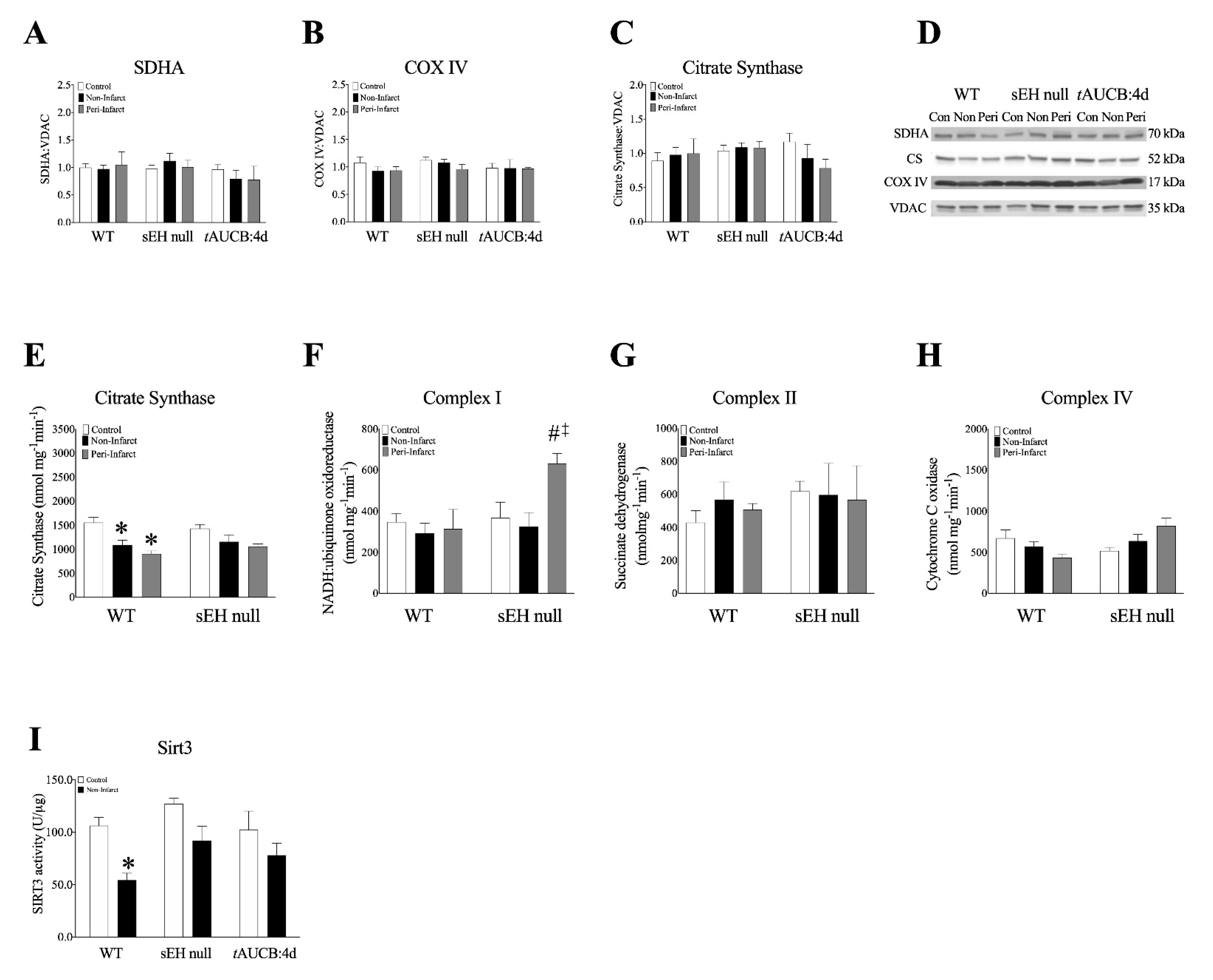
2.6. Decreased Mitochondrial Function in LV Myocardium from ICM Patients
2.7. Human LV Demonstrates Loss of Mitochondrial Ultrastructure and Organization
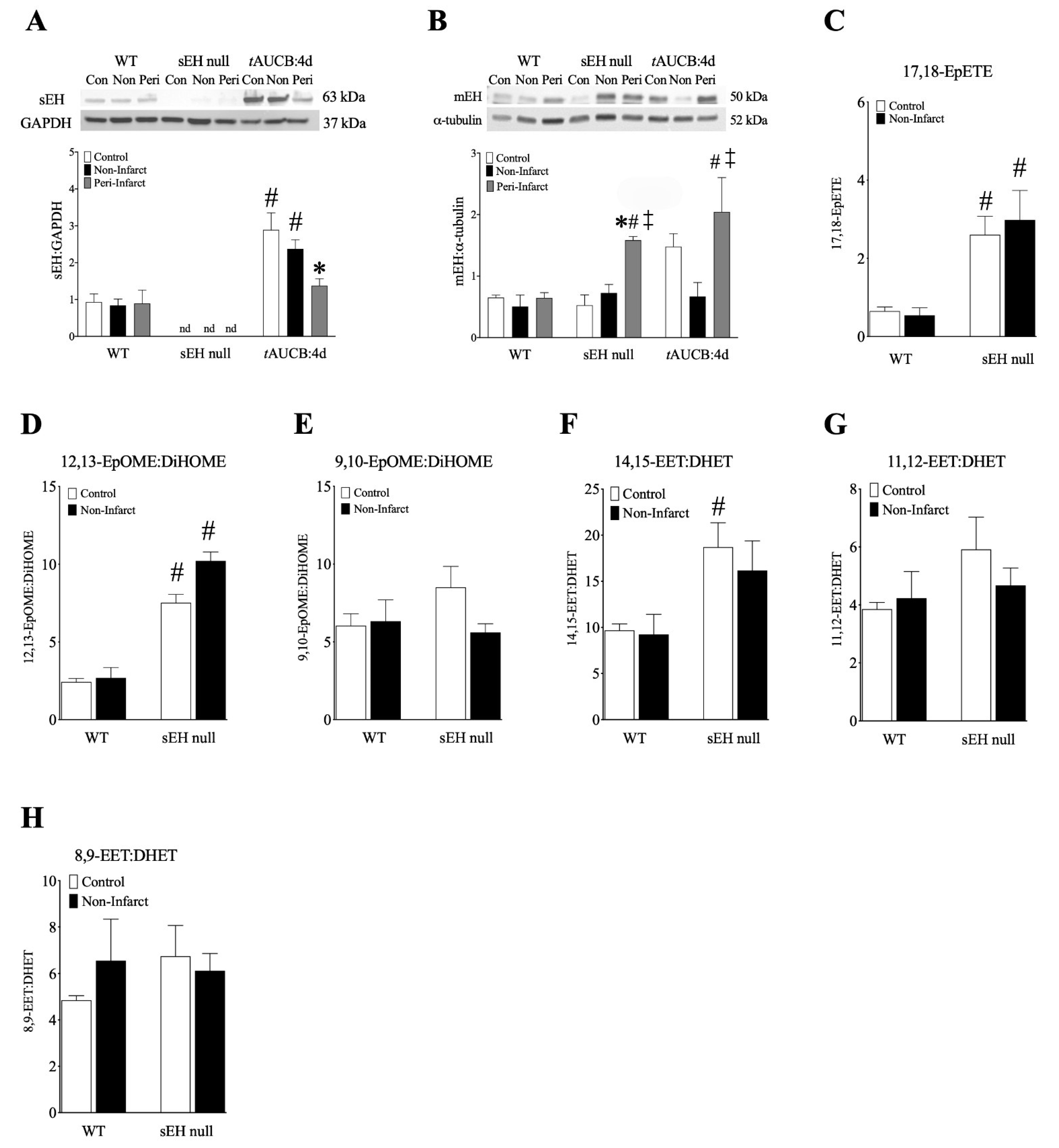
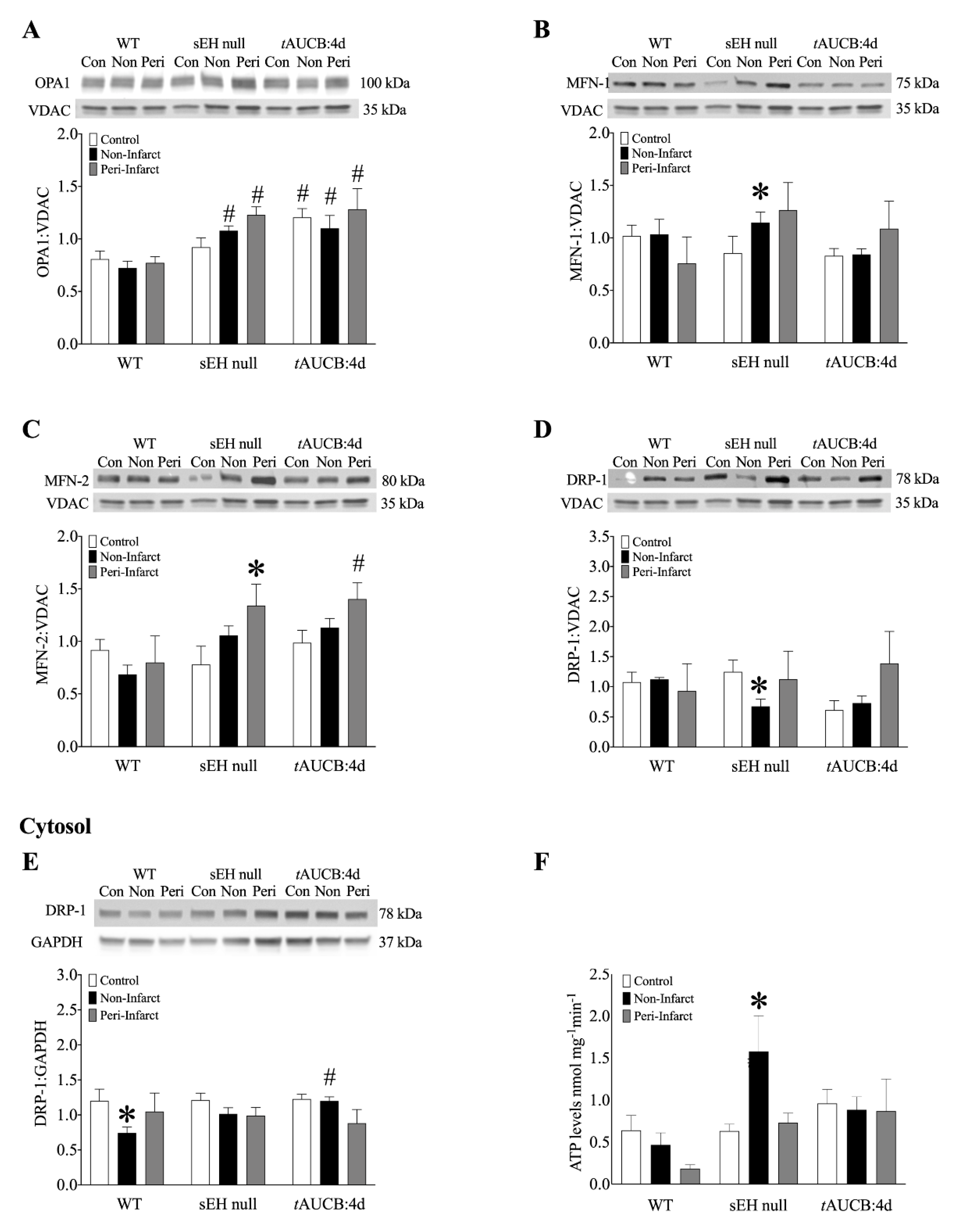
2.8. Female Mice with Genetic Deletion of sEH Demonstrate Improved Protein Expression and Mitochondrial Function Post-MI
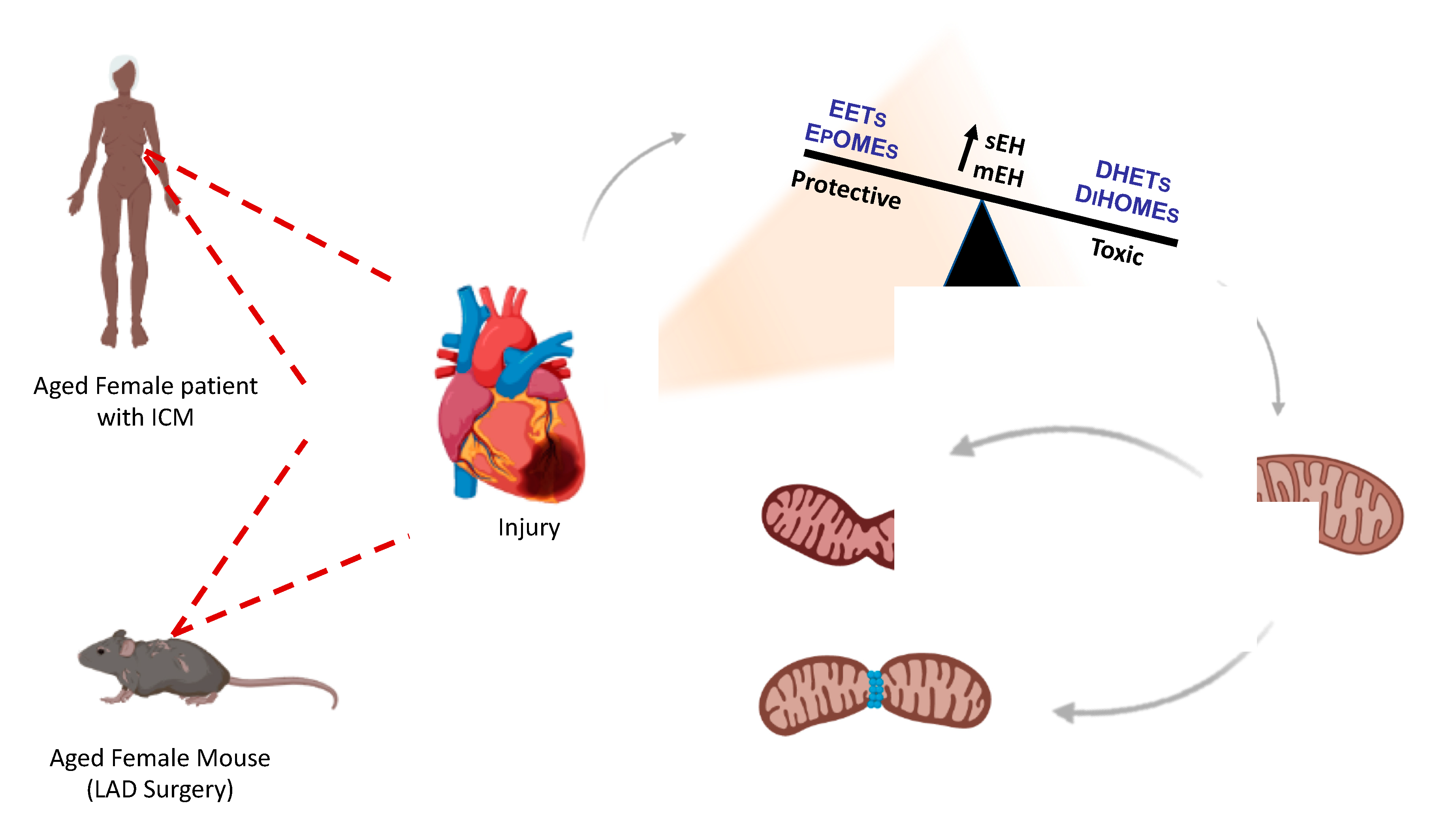
3. Discussion
4. Materials and Methods
4.1. Human Explanted Heart Tissue
4.2. Animal Studies
4.3. Induction of Myocardial Infarction in Mice
4.4. Infarct Assessment
4.5. Mouse Cardiac Function Assessment
4.6. LC-MS/MS
4.7. Protein Immunoblotting
4.8. Mitochondrial Enzyme Activities
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lloyd-Jones, D.M. Slowing Progress in Cardiovascular Mortality Rates: You Reap What You Sow. JAMA Cardiol. 2016, 1, 599–600. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Cheng, S.; Fernandes, V.R.; Bluemke, D.A.; McClelland, R.L.; Kronmal, R.A.; Lima, J.A. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: The Multi-Ethnic Study of Atherosclerosis. Circ. Cardiovasc. Imaging 2009, 2, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Merz, A.A.; Cheng, S. Sex differences in cardiovascular ageing. Heart 2016, 102, 825–831. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; Patel, H.N.; Mehta, L.S.; Sanghani, R.M.; Lundberg, G.P.; Lewis, S.J.; Mendelson, M.A.; Wood, M.J.; Volgman, A.S.; Mieres, J.H. Sex Differences in Ischemic Heart Disease: Advances, Obstacles, and Next Steps. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e004437. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Vaccarino, V. Myocardial Infarction in Young Women. Circulation 2019, 139, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P.S. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010, 106, 1681–1691. [Google Scholar] [CrossRef] [Green Version]
- Ostadal, B.; Drahota, Z.; Houstek, J.; Milerova, M.; Ostadalova, I.; Hlavackova, M.; Kolar, F. Developmental and sex differences in cardiac tolerance to ischemia-reperfusion injury: The role of mitochondria (1). Can. J. Physiol. Pharmacol. 2019, 97, 808–814. [Google Scholar] [CrossRef]
- Parks, R.J.; Howlett, S.E. Sex differences in mechanisms of cardiac excitation-contraction coupling. Pflugers Arch. 2013, 465, 747–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Endo, T.; Darwesh, A.M.; Samokhvalov, V.; Seubert, J.M. Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Age and Sex Differences in Hearts of Soluble Epoxide Hydrolase Null Mice. Front. Physiol. 2020, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Edin, M.L.; Hamedani, B.G.; Gruzdev, A.; Graves, J.P.; Lih, F.B.; Arbes, S.J., 3rd; Singh, R.; Orjuela Leon, A.C.; Bradbury, J.A.; DeGraff, L.M.; et al. Epoxide hydrolase 1 (EPHX1) hydrolyzes epoxyeicosanoids and impairs cardiac recovery after ischemia. J. Biol. Chem. 2018, 293, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhnokh, M.K.; Yang, F.H.; Samokhvalov, V.; Jamieson, K.L.; Cho, W.J.; Wagg, C.; Takawale, A.; Wang, X.; Lopaschuk, G.D.; Hammock, B.D.; et al. Inhibition of Soluble Epoxide Hydrolase Limits Mitochondrial Damage and Preserves Function Following Ischemic Injury. Front. Pharmacol. 2016, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, K.L.; Samokhvalov, V.; Akhnokh, M.K.; Lee, K.; Cho, W.J.; Takawale, A.; Wang, X.; Kassiri, Z.; Seubert, J.M. Genetic deletion of soluble epoxide hydrolase provides cardioprotective responses following myocardial infarction in aged mice. Prostaglandins Lipid Mediat. 2017, 132, 47–58. [Google Scholar] [CrossRef]
- Liu, J.Y.; Tsai, H.J.; Hwang, S.H.; Jones, P.D.; Morisseau, C.; Hammock, B.D. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br. J. Pharmacol. 2009, 156, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillis, G.S.; Moller, J.E.; Pellikka, P.A.; Gersh, B.J.; Wright, R.S.; Ommen, S.R.; Reeder, G.S.; Oh, J.K. Noninvasive estimation of left ventricular filling pressure by E/e’ is a powerful predictor of survival after acute myocardial infarction. J. Am. Coll Cardiol. 2004, 43, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Boukens, B.J.; Rivaud, M.R.; Rentschler, S.; Coronel, R. Misinterpretation of the mouse ECG: ’musing the waves of Mus musculus’. J. Physiol. 2014, 592, 4613–4626. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Ong, S.B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877. [Google Scholar] [CrossRef]
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur. Heart J. 2015, 36, 3404–3412. [Google Scholar] [CrossRef]
- Koentges, C.; Bode, C.; Bugger, H. SIRT3 in Cardiac Physiology and Disease. Front. Cardiovasc. Med. 2016, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.T.; Wasti, B.; Xu, D.Y.; Shen, L.; Du, J.Q.; Zhao, S.P. Soluble epoxide hydrolase and ischemic cardiomyopathy. Int. J. Cardiol. 2012, 155, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; North, K.E.; Bray, M.S.; Fornage, M.; Seubert, J.M.; Newman, J.W.; Hammock, B.D.; Couper, D.J.; Heiss, G.; Zeldin, D.C. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum. Mol. Genet. 2006, 15, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Seubert, J.M.; Sinal, C.J.; Graves, J.; DeGraff, L.M.; Bradbury, J.A.; Lee, C.R.; Goralski, K.; Carey, M.A.; Luria, A.; Newman, J.W.; et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ. Res. 2006, 99, 442–450. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, K.R.; Zordoky, B.N.; Edin, M.L.; Alsaleh, N.; El-Kadi, A.O.; Zeldin, D.C.; Seubert, J.M. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on postischemic cardiac function in aged mice. Prostaglandins Other Lipid Mediat. 2013, 104–105, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannehr, M.; Lohr, L.; Gelep, J.; Haverkamp, W.; Schunck, W.H.; Gollasch, M.; Wutzler, A. Linoleic Acid Metabolite DiHOME Decreases Post-ischemic Cardiac Recovery in Murine Hearts. Cardiovasc. Toxicol. 2019, 19, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Thai, P.N.; Seidlmayer, L.K.; Miller, C.; Ferrero, M.; Dorn, G.W., II; Schaefer, S.; Bers, D.M.; Dedkova, E.N. Mitochondrial Quality Control in Aging and Heart Failure: Influence of Ketone Bodies and Mitofusin-Stabilizing Peptides. Front. Physiol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchu, S.N.; Lee, S.B.; Qadhi, R.S.; Chaudhary, K.R.; El-Sikhry, H.; Kodela, R.; Falck, J.R.; Seubert, J.M. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br. J. Pharmacol. 2011, 162, 897–907. [Google Scholar] [CrossRef] [Green Version]
- El-Sikhry, H.E.; Alsaleh, N.; Dakarapu, R.; Falck, J.R.; Seubert, J.M. Novel Roles of Epoxyeicosanoids in Regulating Cardiac Mitochondria. PLoS ONE 2016, 11, e0160380. [Google Scholar] [CrossRef] [Green Version]
- Samokhvalov, V.; Jamieson, K.L.; Darwesh, A.M.; Keshavarz-Bahaghighat, H.; Lee, T.Y.T.; Edin, M.; Lih, F.; Zeldin, D.C.; Seubert, J.M. Deficiency of Soluble Epoxide Hydrolase Protects Cardiac Function Impaired by LPS-Induced Acute Inflammation. Front. Pharmacol. 2018, 9, 1572. [Google Scholar] [CrossRef] [Green Version]
- Schunck, W.H.; Konkel, A.; Fischer, R.; Weylandt, K.H. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol. Ther. 2018, 183, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, Z.; Shen, M.; Zhang, Y.; Duan, J.; Guo, Y.; Zhang, D.; Hu, J.; Lin, J.; Man, W.; et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1962–1972. [Google Scholar] [CrossRef]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Zeng, W.; Li, L.; Huo, D.; Zeng, L.; Tan, J.; Zhou, J.; Sun, J.; Liu, G.; Li, Y.; et al. PLGA-PNIPAM Microspheres Loaded with the Gastrointestinal Nutrient NaB Ameliorate Cardiac Dysfunction by Activating Sirt3 in Acute Myocardial Infarction. Adv. Sci. 2016, 3, 1600254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Zuloaga, K.L.; Zhang, W.; Roese, N.E.; Alkayed, N.J. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front. Pharmacol. 2014, 5, 290. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchens, M.P.; Nakano, T.; Dunlap, J.; Traystman, R.J.; Hurn, P.D.; Alkayed, N.J. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 2008, 76, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verschoor, C.P.; Tamim, H. Frailty Is Inversely Related to Age at Menopause and Elevated in Women Who Have Had a Hysterectomy: An Analysis of the Canadian Longitudinal Study on Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 675–682. [Google Scholar] [CrossRef]
- Smith, E.R.; Wang, Y.; Xu, X.X. Development of a mouse model of menopausal ovarian cancer. Front. Oncol 2014, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, C.A. Reproductive ageing and the menopause. Int. J. Dev. Biol. 2001, 45, 613–617. [Google Scholar]
- Kandalam, V.; Basu, R.; Abraham, T.; Wang, X.; Soloway, P.D.; Jaworski, D.M.; Oudit, G.Y.; Kassiri, Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ. Res. 2010, 106, 796–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhabyeyev, P.; McLean, B.; Chen, X.; Vanhaesebroeck, B.; Oudit, G.Y. Inhibition of PI3Kinase-alpha is pro-arrhythmic and associated with enhanced late Na(+) current, contractility, and Ca(2+) release in murine hearts. J. Mol. Cell Cardiol. 2019, 132, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | sEH null | WT+tAUCB:4d PreTx | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ECHOCARDIOGRAPHY | Baseline | 7d | 28d | Baseline | 7d | 28d | Baseline | 7d | 28d |
| HR, beats/min | 482 ± 9 | 469 ± 13 | 488 ± 11 | 499 ± 11 | 486 ± 11 | 512 ± 12 | 463 ± 13 | 476 ± 22 | 426 ± 7 |
| Wall measurements | |||||||||
| Corrected LV mass, mg | 111.4 ± 9.8 | 146.3 ± 12.3 | 149.4 ± 9.9 * | 93.9 ± 3.2 | 114.7 ± 7.2 | 134.3 ± 12.4 * | 94.80 ± 5.9 | 132.1 ± 13.5 * | 119.8 ± 14.0 |
| IVS-diastole, mm | 0.89 ± 0.03 | 0.69 ± 0.10 | 0.60 ±0.06 * | 0.87 ± 0.02 | 0.71 ± 0.05 | 0.70 ± 0.07 | 0.78 ± 0.04 | 0.74 ± 0.09 | 0.65 ± 0.17 |
| IVS-systole, mm | 1.32 ± 0.06 | 0.91 ± 0.14 * | 0.76 ± 0.07 * | 1.32 ± 0.04 | 0.91 ± 0.07 * | 0.98 ± 0.10 * | 1.18 ± 0.07 | 1.42 ± 0.20 | 0.78 ± 0.21 |
| LVPW-diastole, mm | 0.90 ± 0.04 | 0.94 ± 0.07 | 0.82 ± 0.06 | 0.85 ± 0.02 | 0.78 ± 0.06 | 0.81 ± 0.05 | 0.81 ± 0.04 | 0.90 ± 0.08 | 0.84 ± 0.05 |
| LVPW-systole, mm | 1.29 ± 0.05 | 1.01 ± 0.09 * | 0.99 ± 0.10 * | 1.25 ± 0.04 | 0.97 ± 0.08 * | 1.04 ± 0.07 | 1.25 ± 0.07 | 1.20 ± 0.11 | 1.14 ± 0.13 |
| LVID-diastole, mm | 3.97 ± 0.13 | 5.10 ± 0.17 * | 5.69 ± 0.16 * | 3.80 ± 0.09 | 4.77 ± 0.21 * | 5.12 ± 0.14 * | 4.04 ± 0.10 | 4.78 ± 0.24 * | 4.87 ± 0.22 |
| LVID-systole, mm | 2.66 ± 0.13 | 4.43 ± 0.28 * | 5.04 ± 0.21 * | 2.43 ± 0.10 | 3.97 ± 0.26 * | 4.34 ± 0.20 * | 2.65 ± 0.11 | 3.76 ± 0.36 * | 3.92 ± 0.29* |
| Cardiac Function, Simpsons | |||||||||
| EF, % | 59.17 ± 1.58 | 27.29 ± 1.38 * | 23.13 ± 3.76 * | 65.29 ± 1.53 | 37.05 ± 2.44 *# | 30.25 ± 3.03 * | 61.00 ± 2.99 | 41.91 ± 5.00 *# | 33.53 ± 5.15 * |
| FAC, % | 48.86 ± 2.38 | 17.96 ± 2.87 * | 16.63± 3.04 * | 55.92 ± 2.44 | 29.34 ± 3.10 *# | 22.94 ± 2.06 * | 53.63 ± 3.27 | 31.75 ± 5.61 *# | 29.55 ± 2.61 * |
| LVEDV, µL | 75.76 ± 4.20 | 140.86 ± 16.13 * | 179.78 ± 19.63 * | 66.74 ± 2.85 | 116.31 ± 10.40 * | 138.18 ± 11.23 * | 64.65 ± 6.15 | 108.39 ± 17.54 * | 114.53 ± 9.42 * |
| LVESV, µL | 31.37 ± 2.56 | 103.20 ± 12.78 * | 143.07 ± 20.50 * | 23.44 ± 1.72 | 75.87 ± 9.19 * | 99.70 ± 11.31 *# | 25.50 ± 4.08 | 71.47 ± 16.23 * | 69.10 ± 10.20 * |
| CO, mL/min | 21.31 ± 1.00 | 17.51 ± 1.89 | 16.86 ± 1.64 | 21.96 ± 0.84 | 20.32 ± 1.17 | 19.57 ± 1.41 | 18.18 ± 1.14 | 17.91 ± 2.25 | 22.82 ± 3.01 |
| SV, µL | 44.39 ± 2.09 | 37.66 ± 3.67 | 35.97 ± 3.41 | 43.59 ± 1.71 | 40.44 ± 1.95 | 38.48 ± 2.97 | 39.15 ± 2.71 | 36.92 ± 3.87 | 45.44 ± 5.02 |
| Doppler Imaging | |||||||||
| IVRT, ms | 16.02 ± 0.78 | 18.74 ± 2.07 | 19.95 ± 1.94 | 15.27 ± 0.94 | 16.77 ± 0.91 | 25.67 ± 4.57 *‡ | 16.27 ± 0.79 | 19.91 ± 1.09 | 21.87 ± 2.81 |
| IVCT, ms | 14.96 ± 1.16 | 19.64 ± 2.75 | 31.28 ± 7.58 *‡ | 11.12 ± 0.64 | 20.99 ± 3.04 * | 18.99 ± 3.50 # | 15.22 ± 0.96 | 23.17 ± 2.73 | 26.31 ± 0.78 |
| ET, ms | 40.77 ± 1.45 | 36.51 ± 2.35 | 39.22 ± 2.37 | 39.71 ± 0.89 | 38.47 ± 1.57 | 35.60 ± 2.08 | 46.55 ± 1.14 # | 42.79 ± 0.96 # | 51.96 ± 6.38 # |
| Tei index | 0.76 ± 0.03 | 1.09 ± 0.08 * | 1.25 ± 0.11 * | 0.67 ± 0.03 | 1.00 ± 0.13 * | 1.24 ± 0.20 * | 0.68 ± 0.01 | 1.01 ± 0.08 * | 0.94 ± 0.04 |
| E’ | 23.71 ± 1.48 | 22.24 ± 4.50 | 13.61 ± 3.01 * | 25.85 ± 1.50 | 18.51 ± 1.74 * | 16.54 ± 2.47 * | 27.72 ± 1.96 | 19.32 ± 1.26 * | 25.38 ± 10.87 |
| E’/A’ | 1.15 ± 0.05 | 1.28 ± 0.25 | 1.22 ± 0.32 | 1.32 ± 0.13 | 1.25 ± 0.11 | 0.98 ± 0.10 | 1.35 ± 0.08 | 1.68 ± 0.32 | 0.96 ± 0.14 |
| E/E’ | 22.53 ± 1.23 | 32.16 ± 6.37 | 45.23 ± 7.72 * | 23.05 ± 1.36 | 34.47 ± 4.93 * | 41.50 ± 4.69 * | 21.26 ± 1.84 | 29.83 ± 4.61 | 33.64 ± 12.42 |
| ELECTROCARDIOGRAM | |||||||||
| HR, beats/min | 471 ± 14 | 488 ± 11 | 523 ± 5 | 505 ± 21 | 475 ± 31 | 529 ± 14 | |||
| RR, ms | 128.1 ± 3.8 | 123.3 ± 2.9 | 114.8 ± 1.0 | 121.6 ± 4.3 | 129.4 ± 8.2 | 112.7 ± 4.0 | |||
| QRS, ms | 12.0 ± 0.3 | 10.7 ± 0.2 | 15.1 ± 1.7 *‡ | 11.7 ± 0.2 | 10.3 ± 0.4 | 10.9 ± 1.6 # | |||
| PR, ms | 43.3 ± 1.5 | 40.8 ± 1.6 | 51.8 ± 3.2 *‡ | 42.1 ± 1.0 | 42.0 ± 0.3 | 44.6 ± 1.4 # | |||
| QTcF, ms | 45.7 ± 0.8 | 74.0 ± 2.2 * | 65.8 ± 7.6 * | 45.1 ± 1.0 | 70.2 ± 5.1 * | 58.5 ± 1.1 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamieson, K.L.; Darwesh, A.M.; Sosnowski, D.K.; Zhang, H.; Shah, S.; Zhabyeyev, P.; Yang, J.; Hammock, B.D.; Edin, M.L.; Zeldin, D.C.; et al. Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury. Int. J. Mol. Sci. 2021, 22, 1691. https://doi.org/10.3390/ijms22041691
Jamieson KL, Darwesh AM, Sosnowski DK, Zhang H, Shah S, Zhabyeyev P, Yang J, Hammock BD, Edin ML, Zeldin DC, et al. Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury. International Journal of Molecular Sciences. 2021; 22(4):1691. https://doi.org/10.3390/ijms22041691
Chicago/Turabian StyleJamieson, K. Lockhart, Ahmed M. Darwesh, Deanna K. Sosnowski, Hao Zhang, Saumya Shah, Pavel Zhabyeyev, Jun Yang, Bruce D. Hammock, Matthew L. Edin, Darryl C. Zeldin, and et al. 2021. "Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury" International Journal of Molecular Sciences 22, no. 4: 1691. https://doi.org/10.3390/ijms22041691
APA StyleJamieson, K. L., Darwesh, A. M., Sosnowski, D. K., Zhang, H., Shah, S., Zhabyeyev, P., Yang, J., Hammock, B. D., Edin, M. L., Zeldin, D. C., Oudit, G. Y., Kassiri, Z., & Seubert, J. M. (2021). Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury. International Journal of Molecular Sciences, 22(4), 1691. https://doi.org/10.3390/ijms22041691