
PEG-Coated Large Mesoporous Silicas as Smart Platform for Protein Delivery and Their Use in a Collagen-Based Formulation for 3D Printing

 ,
,  ,
, 

Abstract
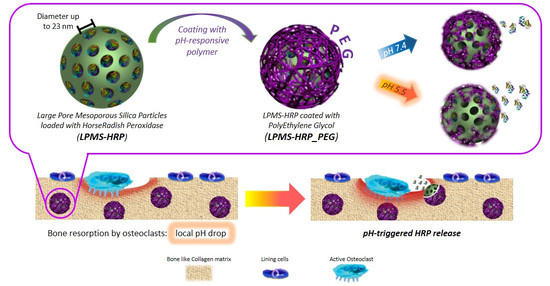
:
1. Introduction
2. Materials and Methods
2.1. Preparation of Large-Pore Mesoporous Silica Particles
2.1.1. Synthesis of Mesoporous Silica-Based Particles with Large Pores (LPMSs)
2.1.2. Characterization of LPMSs
2.2. Study of Protein Adsorption and Release
2.2.1. Procedure for HRP Adsorption into LPMS_140_24 Mesopores (LPMS-HRP)
2.2.2. Evaluation of the Effective HRP Adsorption
2.2.3. In Vitro Protein Release Test
2.3. pH-Responsive Surface Coating of LPMS-HRP
2.3.1. Preparation and Characterization of PEG-Coated LPMS-HRP via PEGylation (LPMS-HRP_PEG)
2.3.2. In Vitro HRP Release Kinetics at pH 5.5 and pH 7.4 from LPMS-HRP_PEG Carriers
2.4. 3D Printing of the Collagen-Based Composite Systems
2.4.1. Preparation of the Composite Suspension Based on Collagen and LPMS-HRP_PEG
2.4.2. Rheological Characterization
2.4.3. 3D Printing of 3D Mesh-Like Composite Scaffolds
3. Results and Discussion
3.1. LPMS Particles Characterization
3.2. HRP Absorption Efficiency Assessment and In Vitro Release Test
3.3. PEG Coating Assessment and In Vitro Evaluation of the Triggering Release Kinetics in Acidic Conditions
3.4. 3D Printing of the Collagen-Based Composite System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Witharana, C.; Wanigasekara, J. Drug Delivery Systems: A New Frontier in Nano-Technology. Int. J. Med. Res. Health Sci. 2017, 6, 11–14. [Google Scholar]
- Tiwari, G.; Tiwari, R.; Bannerjee, S.; Bhati, L.; Pandey, S.; Pandey, P.; Sriwastawa, B. Drug Delivery Systems: An Updated Review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, J. Controlled Release of Biologically Active Compounds from Bioerodible Polymers. Biomater. Silver Jubil. Compend. 1980, 1–7. [Google Scholar] [CrossRef]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Zhou, Y.; Fan, T.; Lin, Y.; Zhang, H.; Mei, L. Inorganic Nano-Carriers Based Smart Drug Delivery Systems for Tumor Therapy. Smart Mater. Med. 2020, 1, 32–47. [Google Scholar] [CrossRef]
- Quignard, S.; Coradin, T.; Powell, J.J.; Jugdaohsingh, R. Silica Nanoparticles as Sources of Silicic Acid Favoring Wound Healing in Vitro. Colloids Surf. B Biointerfaces 2017, 155, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamam, F.; Nasr, A. Curcumin-Loaded Mesoporous Silica Particles as Wound-Healing Agent: An In Vivo Study. Saudi J. Med. Med. Sci. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Zhang, K.R.; Gao, H.L.; Zhou, Y.; Yan, B.B.; Yang, C.; Zhang, Z.Y.; Dong, L.; Chen, S.M.; Xu, R.; et al. Activating Proper Inflammation for Wound-Healing Acceleration via Mesoporous Silica Nanoparticle Tissue Adhesive. Nano Res. 2020, 13, 373–379. [Google Scholar] [CrossRef]
- Iturrioz-Rodríguez, N.; Correa-Duarte, M.A.; Fanarraga, M.L. Controlled Drug Delivery Systems for Cancer Based on Mesoporous Silica Nanoparticles. Int. J. Nanomed. 2019, 14, 3389–3401. [Google Scholar] [CrossRef] [Green Version]
- Arap, W.; Pasqualini, R.; Montalti, M.; Petrizza, L.; Prodi, L.; Rampazzo, E.; Zaccheroni, N.; Marchiò, S. Luminescent Silica Nanoparticles for Cancer Diagnosis. Curr. Med. Chem. 2013, 20, 2195–2211. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Huxford-Phillips, R.C.; Lin, W. Silica-Based Nanoprobes for Biomedical Imaging and Theranostic Applications. Chem. Soc. Rev. 2012, 41, 2673–2685. [Google Scholar] [CrossRef] [Green Version]
- Shirshahi, V.; Soltani, M. Solid Silica Nanoparticles: Applications in Molecular Imaging. Contrast Media Mol. Imaging 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Colilla, M.; Vallet-Regí, M. Nanostructured Mesoporous Silicas for Bone Tissue Regeneration. J. Nanomater. 2008, 2008. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, X.; He, C. Mesoporous Silica Nanoparticles for Tissue-Engineering Applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11. [Google Scholar] [CrossRef]
- Shadjou, N.; Hasanzadeh, M. Silica-Based Mesoporous Nanobiomaterials as Promoter of Bone Regeneration Process. J. Biomed. Mater. Res. Part. A 2015, 103, 3703–3716. [Google Scholar] [CrossRef]
- Gisbert-Garzarán, M.; Manzano, M.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for the Treatment of Complex Bone Diseases: Bone Cancer, Bone Infection and Osteoporosis. Pharmaceutics 2020, 12, 83. [Google Scholar] [CrossRef] [Green Version]
- Bitar, A.; Ahmad, N.M.; Fessi, H.; Elaissari, A. Silica-Based Nanoparticles for Biomedical Applications. Drug Discov. Today 2012, 17, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Quan, G.; Wu, Q.; Zhang, X.; Niu, B.; Wu, B.; Huang, Y.; Pan, X.; Wu, C. Mesoporous Silica Nanoparticles for Drug and Gene Delivery. Acta Pharm. Sin. B 2018, 8, 165–177. [Google Scholar] [CrossRef]
- Knežević, N.; Durand, J.O. Large Pore Mesoporous Silica Nanomaterials for Application in Delivery of Biomolecules. Nanoscale 2015, 7, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Derakhshankhah, H.; Alaei, L.; Fattahi, A.; Varnamkhasti, B.S.; Saboury, A.A. Mesoporous Silica Nanoparticles for Therapeutic/Diagnostic Applications. Biomed. Pharmacother. 2019, 109, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Chang, J. Well-Ordered Mesoporous Bioactive Glasses (MBG): A Promising Bioactive Drug Delivery System. J. Control. Release 2006, 110, 522–530. [Google Scholar] [CrossRef]
- Labouta, H.I.; Schneider, M. Interaction of Inorganic Nanoparticles with the Skin Barrier: Current Status and Critical Review. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 39–54. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Rámila, A.; Del Real, R.P.; Pérez-Pariente, J. A New Property of MCM-41: Drug Delivery System. Chem. Mater. 2001, 13, 308–311. [Google Scholar] [CrossRef]
- Juère, E.; Kleitz, F. On the Nanopore Confinement of Therapeutic Drugs into Mesoporous Silica Materials and Its Implications. Microporous Mesoporous Mater. 2018, 270, 109–119. [Google Scholar] [CrossRef]
- Farjadian, F.; Roointan, A.; Mohammadi-Samani, S.; Hosseini, M. Mesoporous Silica Nanoparticles: Synthesis, Pharmaceutical Applications, Biodistribution, and Biosafety Assessment. Chem. Eng. J. 2019, 359, 684–705. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Lindén, M. Towards Establishing Structure-Activity Relationships for Mesoporous Silica in Drug Delivery Applications. J. Control. Release 2008, 128, 157–164. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Vallet-Regi, M. Mesoporous Bioactive Glasses: Relevance of Their Porous Structure Compared to That of Classical Bioglasses. Biomed. Glas. 2015, 1, 140–150. [Google Scholar] [CrossRef]
- Niu, D.; Ma, Z.; Li, Y.; Shi, J. Synthesis of Core-Shell Structured Dual-Mesoporous Silica Spheres with Tunable Pore Size and Controllable Shell Thickness. J. Am. Chem. Soc. 2010, 132, 15144–15147. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, P.; Chen, H.; Li, Y.; Bu, W.; Shu, Z.; Li, Y.; Zhang, J.; Zhang, L.; Pan, L.; et al. Colloidal HPMO Nanoparticles: Silica-Etching Chemistry Tailoring, Topological Transformation, and Nano-Biomedical Applications. Adv. Mater. 2013, 25, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Na, H.K.; Kim, Y.K.; Ryoo, S.R.; Cho, H.S.; Lee, K.E.; Jeon, H.; Ryoo, R.; Min, D.H. Facile Synthesis of Monodispersed Mesoporous Silica Nanoparticles with Ultralarge Pores and Their Application in Gene Delivery. ACS Nano 2011, 5, 3568–3576. [Google Scholar] [CrossRef]
- Lee, C.H.; Lin, T.S.; Mou, C.Y. Mesoporous Materials for Encapsulating Enzymes. Nano Today 2009, 4, 165–179. [Google Scholar] [CrossRef]
- Carlsson, N.; Gustafsson, H.; Thörn, C.; Olsson, L.; Holmberg, K.; Åkerman, B. Enzymes Immobilized in Mesoporous Silica: A Physical-Chemical Perspective. Adv. Colloid Interface Sci. 2014, 205, 339–360. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.B.; Lee, J.W.; Shim, W.G.; Moon, H. Synthesis of Functionalized SBA-15 with Ordered Large Pore Size and Its Adsorption Properties of BSA. Microporous Mesoporous Mater. 2008, 110, 560–569. [Google Scholar] [CrossRef]
- Kruk, M. Access to Ultralarge-Pore Ordered Mesoporous Materials through Selection of Surfactant/Swelling-Agent Micellar Templates. Acc. Chem. Res. 2012, 45, 1678–1687. [Google Scholar] [CrossRef]
- Schmidt-winkel, P.; Lukens, W.W.; Zhao, D.; Yang, P.; Chmelka, B.F.; Stucky, G.D.; Barbara, S. Mesocellular Siliceous Foams with Uniformly Sized Cells and Windows Molecular Sieves with Uniform Large Pores Are Desirable for Chemical Reactions and for Use in Separations Involving Large Molecules. 1 Periodic Cubic and Hexagonal Mesoporous Silica Phas. J. Am. Chem. Soc. 2000, 121, 254–255. [Google Scholar] [CrossRef]
- Ma, J.; Liu, Q.; Chen, D.; Wen, S.; Wang, T. Synthesis and Characterisation of Pore-Expanded Mesoporous Silica Materials. Micro Nano Lett. 2015, 10, 140–144. [Google Scholar] [CrossRef]
- Blin, J.L.; Su, B.L. Tailoring Pore Size of Ordered Mesoporous Silicas Using One or Two Organic Auxiliaries as Expanders. Langmuir 2002, 18, 5303–5308. [Google Scholar] [CrossRef]
- Xin, C.; Zhao, N.; Zhan, H.; Xiao, F.; Wei, W.; Sun, Y. Phase Transition of Silica in the TMB-P123-H2O-TEOS Quadru-Component System: A Feasible Route to Different Mesostructured Materials. J. Colloid Interface Sci. 2014, 433, 176–182. [Google Scholar] [CrossRef]
- Wei, J.; Wang, H.; Deng, Y.; Sun, Z.; Shi, L.; Tu, B.; Luqman, M.; Zhao, D. Solvent Evaporation Induced Aggregating Assembly Approach to Three-Dimensional Ordered Mesoporous Silica with Ultralarge Accessible Mesopores. J. Am. Chem. Soc. 2011, 133, 20369–20377. [Google Scholar] [CrossRef]
- Huang, L.; Kruk, M. Synthesis of Ultra-Large-Pore FDU-12 Silica Using Ethylbenzene as Micelle Expander. J. Colloid Interface Sci. 2012, 365, 137–142. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, J.; Ma, D.; Bao, X.; Klein-Hoffmann, A.; Weinberg, G.; Su, D.; Schlögl, R. Unusual Mesoporous SBA-15 with Parallel Channels Running along the Short Axis. J. Am. Chem. Soc. 2004, 126, 7440–7441. [Google Scholar] [CrossRef]
- Egger, S.M.; Hurley, K.R.; Datt, A.; Swindlehurst, G.; Haynes, C.L. Ultraporous Mesostructured Silica Nanoparticles. Chem. Mater. 2015, 27, 3193–3196. [Google Scholar] [CrossRef]
- Blin, J.L.; Otjacques, C.; Herrier, G.; Su, B.L. Pore Size Engineering of Mesoporous Silicas Using Decane as Expander. Langmuir 2000, 16, 4229–4236. [Google Scholar] [CrossRef]
- Shan, W.; Wang, W.; Ru, H. Siliceous Mesocellular Foams Modified via a Partitioned Cooperative Self-Assembly Process Using Hexane as Pore Swelling Agent. J. Non. Cryst. Solids 2015, 425, 183–189. [Google Scholar] [CrossRef]
- Boahene, P.E.; Soni, K.K.; Dalai, A.K.; Adjaye, J. Application of Different Pore Diameter SBA-15 Supports for Heavy Gas Oil Hydrotreatment Using FeW Catalyst. Appl. Catal. A Gen. 2011, 402, 31–40. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, H.; Ma, D.; Chen, Y.; Bao, X.; Klein-Hoffmann, A.; Pfänder, N.; Su, D.S. Alkanes-Assisted Low Temperature Formation of Highly Ordered SBA-15 with Large Cylindrical Mesopores. Chem. Commun. 2005, 5343–5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Kruk, M. Synthesis of Large-Pore SBA-15 Silica from Tetramethyl Orthosilicate Using Triisopropylbenzene as Micelle Expander. Colloids Surf. A Physicochem. Eng. Asp. 2010, 357, 91–96. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, H.; Tian, R.; Ma, D.; Bao, X.; Su, D.S.; Zou, H. Ultrafast Enzyme Immobilization over Large-Pore Nanoscale Mesoporous Silica Particles. Chem. Commun. 2006, 1, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Bu, X.; Pine, D.J. Control of Pore Sizes in Mesoporous Silica Templated by Liquid Crystals in Block Copolymer-Cosurfactant-Water Systems. Langmuir 2000, 16, 5304–5310. [Google Scholar] [CrossRef]
- Man, T. Design of Well-Defined Mesoporous Silicas via Surfactant Templating Method Enhanced by the Use of Swelling Agents; City University of New York (CUNY): New York, NY, USA, 2014; Available online: https://academicworks.cuny.edu/gc_etds/492 (accessed on 5 February 2021).
- Lawrence, G.; Baskar, A.V.; El-Newehy, M.H.; Cha, W.S.; Al-Deyab, S.S.; Vinu, A. Quick High-Temperature Hydrothermal Synthesis of Mesoporous Materials with 3D Cubic Structure for the Adsorption of Lysozyme. Sci. Technol. Adv. Mater. 2015, 16, 24806. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Shin, Y.; Liu, J.; Ackerman, E.J. Entrapping Enzyme in a Functionalized Nanoporous Support. J. Am. Chem. Soc. 2002, 124, 11242–11243. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Lee, K.C.; Kwon, Y.U. Nanoparticle Routes to Mesoporous Titania Thin Films. Chem. Commun. 2001, 1, 1738–1739. [Google Scholar] [CrossRef]
- Aksay, I.A.; Trau, M.; Manne, S.; Honma, I.; Yao, N.; Zhou, L.; Fenter, P.; Eisenberger, P.M.; Gruner, S.M. Biomimetic Pathways for Assembling Inorganic Thin Films. Science 1996, 273, 892–898. [Google Scholar] [CrossRef]
- Borciani, G.; Montalbano, G.; Baldini, N.; Cerqueni, G.; Vitale-Brovarone, C.; Ciapetti, G. Co–Culture Systems of Osteoblasts and Osteoclasts: Simulating in Vitro Bone Remodeling in Regenerative Approaches. Acta Biomater. 2020, 108, 22–45. [Google Scholar] [CrossRef] [PubMed]
- Licini, C.; Vitale-Brovarone, C.; Mattioli-Belmonte, M. Collagen and Non-Collagenous Proteins Molecular Crosstalk in the Pathophysiology of Osteoporosis. Cytokine Growth Factor Rev. 2019, 49, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci. 2018, 19, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, M.D.; Jay, M.; Dziubla, T.D.; Lu, X. PEGylation of Nanocarrier Drug Delivery Systems: State of the Art. J. Biomed. Nanotechnol. 2008, 4, 133–148. [Google Scholar] [CrossRef]
- Joralemon, M.J.; McRae, S.; Emrick, T. PEGylated Polymers for Medicine: From Conjugation to Self-Assembled Systems. Chem. Commun. 2010, 46, 1377–1393. [Google Scholar] [CrossRef] [Green Version]
- D’souza, A.A.; Shegokar, R. Polyethylene Glycol (PEG): A Versatile Polymer for Pharmaceutical Applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef]
- Jang, H.-J.; Shin, C.Y.; Kim, K.-B. Safety Evaluation of Polyethylene Glycol (PEG) Compounds for Cosmetic Use. Toxicol. Res. 2015, 31, 105–136. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Koide, T.; Hyon, S.H. Tribological Characteristics of Polyethylene Glycol (PEG) as a Lubricant for Wear Resistance of Ultra-High-Molecular-Weight Polyethylene (UHMWPE) in Artificial Knee Join. J. Mech. Behav. Biomed. Mater. 2014, 38, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Luo, Y.; Xiang, P.; Yang, X.; Shen, M. Analysis of PEG Oligomers in Black Gel Inks: Discrimination and Ink Dating. Forensic Sci. Int. 2017, 277, 1–9. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(Ethylene Glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Dombb, A.; Miillerd, R.H.; Langerf, R.; Gref, R.; Domb, A.; Quellec, P.; Blunk, T.; Müller, R.H.; Verbavatz, J.M.; Langer, R. The Controlled Intravenous Delivery of Drugs Using PEG-Coated Sterically Stabilized Nanospheres. Adv. Drug Deliv. Rev. 1995, 16, 215–233. [Google Scholar]
- Bunker, A. Poly (Ethylene Glycol) in Drug Delivery, Why Does It Work, and Can We Do Better? All Atom Molecular Dynamics Simulation Provides Some Answers. Phys. Procedia 2012, 34, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Fam, S.Y.; Chee, C.F.; Yong, C.Y.; Ho, K.L.; Mariatulqabtiah, A.R.; Tan, W.S. Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials 2020, 10, 787. [Google Scholar] [CrossRef] [Green Version]
- Dhivya, R.; Ranjani, J.; Bowen, P.K.; Rajendhran, J.; Mayandi, J.; Annaraj, J. Biocompatible Curcumin Loaded PMMA-PEG/ZnO Nanocomposite Induce Apoptosis and Cytotoxicity in Human Gastric Cancer Cells. Mater. Sci. Eng. C 2017, 80, 59–68. [Google Scholar] [CrossRef]
- Stillman, Z.; Jarai, B.M.; Raman, N.; Patel, P.; Fromen, C.A. Degradation Profiles of Poly (Ethylene Glycol)Diacrylate (PEGDA)-Based Hydrogel Nanoparticles. Polym. Chem. 2020, 11, 568–580. [Google Scholar] [CrossRef]
- Gidi, Y.; Bayram, S.; Ablenas, C.J.; Blum, A.S.; Cosa, G. Efficient One-Step PEG-Silane Passivation of Glass Surfaces for Single-Molecule Fluorescence Studies. ACS Appl. Mater. Interfaces 2018, 10, 39505–39511. [Google Scholar] [CrossRef]
- Montalbano, G.; Fiorilli, S.; Caneschi, A.; Vitale-Brovarone, C. Type I Collagen and Strontium-Containing Mesoporous Glass Particles as Hybrid Material for 3D Printing of Bone-like Materials. Materials 2018, 11, 700. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, G.; Borciani, G.; Pontremoli, C.; Ciapetti, G.; Mattioli-Belmonte, M.; Fiorilli, S.; Vitale-Brovarone, C. Development and Biocompatibility of Collagen-Based Composites Enriched with Nanoparticles of Strontium Containing Mesoporous Glass. Materials 2019, 12, 3719. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, G.; Molino, G.; Fiorilli, S.; Vitale-Brovarone, C. Synthesis and Incorporation of Rod-like Nano-Hydroxyapatite into Type I Collagen Matrix: A Hybrid Formulation for 3D Printing of Bone Scaffolds. J. Eur. Ceram. Soc. 2020, 40, 3689–3697. [Google Scholar] [CrossRef]
- Montalbano, G.; Borciani, G.; Cerqueni, G.; Licini, C.; Banche-Niclot, F.; Janner, D.; Sola, S.; Fiorilli, S.; Mattioli-Belmonte, M.; Ciapetti, G.; et al. Collagen Hybrid Formulations for the 3D Printing of Nanostructured Bone Scaffolds: An Optimized Genipin-Crosslinking Strategy. Nanomaterials 2020, 10, 1681. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Yan, X.; Li, Y.; Xiao, L.; Huang, Z.; Lu, Y.; Fan, J. Ordered Nanoporous Silica with Periodic 30-60 Nm Pores as an Effective Support for Gold Nanoparticle Catalysts with Enhanced Lifetime. J. Am. Chem. Soc. 2010, 132, 9596–9597. [Google Scholar] [CrossRef] [PubMed]
- Gajhede, M.; Schuller, D.J.; Henriksen, A.; Smith, A.T.; Poulos, T.L. Crystal Structure of Horseradish Peroxidase C at 2.15 Å Resolution. Nat. Struct. Biol. 1997, 4, 1032–1038. [Google Scholar] [CrossRef]
- Chouyyok, W.; Panpranot, J.; Thanachayanant, C.; Prichanont, S. Effects of PH and Pore Characters of Mesoporous Silicas on Horseradish Peroxidase Immobilization. J. Mol. Catal. B Enzym. 2009, 56, 246–252. [Google Scholar] [CrossRef]
- Tu, J.; Boyle, A.L.; Friedrich, H.; Bomans, P.H.H.; Bussmann, J.; Sommerdijk, N.A.J.M.; Jiskoot, W.; Kros, A. Mesoporous Silica Nanoparticles with Large Pores for the Encapsulation and Release of Proteins. ACS Appl. Mater. Interfaces 2016, 8, 32211–32219. [Google Scholar] [CrossRef]
- Totovao, R. Stimuli-Responsive Breakable Hybrid Organic/Inorganic Silica Nanoparticles for Biomedical Applications. Ph.D. Thesis, Université de Strasbourg, Strasbourg, France, 2017. [Google Scholar]
- Schwarcz, H.P.; Abueidda, D.; Jasiuk, I. The Ultrastructure of Bone and Its Relevance to Mechanical Properties. Front. Phys. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Orban, J.M.; Wilson, L.B.; Kofroth, J.A.; El-Kurdi, M.S.; Maul, T.M.; Vorp, D.A. Crosslinking of Collagen Gels by Transglutaminase. J. Biomed. Mater. Res. Part. A 2004, 68, 756–762. [Google Scholar] [CrossRef]
- Fortunati, D.; Chau, D.Y.S.; Wang, Z.; Collighan, R.J.; Griffin, M. Cross-Linking of Collagen I by Tissue Transglutaminase Provides a Promising Biomaterial for Promoting Bone Healing. Amino Acids 2014, 46, 1751–1761. [Google Scholar] [CrossRef]
- Chau, D.Y.S.; Collighan, R.J.; Verderio, E.A.M.; Addy, V.L.; Griffin, M. The Cellular Response to Transglutaminase-Cross-Linked Collagen. Biomaterials 2005, 26, 6518–6529. [Google Scholar] [CrossRef]
- Hinton, T.J.; Jallerat, Q.; Palchesko, R.N.; Park, J.H.; Grodzicki, M.S.; Shue, H.J.; Ramadan, M.H.; Hudson, A.R.; Feinberg, A.W. Three-Dimensional Printing of Complex Biological Structures by Freeform Reversible Embedding of Suspended Hydrogels. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Dou, Q.; Karim, A.A.; Loh, X.J. Modification of Thermal and Mechanical Properties of PEG-PPG-PEG Copolymer (F127) with MA-POSS. Polymers 2016, 8, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Lie, J.; Wang, L.; Yu, C.; Tu, B.; Zhao, D. Rapid and High-Capacity Immobilization of Enzymes Based on Mesoporous Silicas with Controlled Morphologies. Chem. Commun. 2003, 3, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Washmon-Kriel, L.; Jimenez, V.L.; Balkus, K.J. Cytochrome c Immobilization into Mesoporous Molecular Sieves. J. Mol. Catal. B Enzym. 2000, 10, 453–469. [Google Scholar] [CrossRef]
- Mazinani, B.; Beitollahi, A.; Masrom, A.K.; Ibrahim, S.; Jamil, F. The Effect of Aging Temperature on the Pores of Mesoporous SBA-15 Silica. AIP Conf. Proc. 2012, 1502, 272–279. [Google Scholar] [CrossRef]
- Song, H.M.; Zink, J.I. Ag(i)-Mediated Self-Assembly of Anisotropic Rods and Plates in the Surfactant Mixture of CTAB and Pluronics. RSC Adv. 2019, 9, 4380–4389. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, K.; Pedersen, J.S. Structural Study on the Micelle Formation of Poly (Ethylene Oxide)–Poly (Propylene Oxide)–Poly(Ethylene Oxide) Triblock Copolymer in Aqueous Solution. Macromolecules 1993, 26, 805–812. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Nocera, P.; Piccolella, S.; Pacifico, S. Entrapping Quercetin in Silica/Polyethylene Glycol Hybrid Materials: Chemical Characterization and Biocompatibility. Mater. Sci. Eng. C 2016, 68, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Seker, A.; Arslan, B.; Chen, S. Recovery of Polyphenols from Grape Pomace Using Polyethylene Glycol (PEG)-Grafted Silica Particles and PEG-Assisted Cosolvent Elution. Molecules 2019, 24, 2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paxton, N.; Smolan, W.; Böck, T.; Melchels, F.; Groll, J.; Jungst, T. Proposal to Assess Printability of Bioinks for Extrusion-Based Bioprinting and Evaluation of Rheological Properties Governing Bioprintability. Biofabrication 2017, 9, 44107. [Google Scholar] [CrossRef]
- Schwab, A.; Levato, R.; D’Este, M.; Piluso, S.; Eglin, D.; Malda, J. Printability and Shape Fidelity of Bioinks in 3D Bioprinting. Chem. Rev. 2020, 120, 11028–11055. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous Silica Nanoparticles: Synthesis, Biocompatibility and Drug Delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef]
- Alcantar, N.A.; Aydil, E.S.; Israelachvili, J.N. Polyethylene Glycol-Coated Biocompatible Surfaces. J. Biomed. Mater. Res. 2000, 51, 343–351. [Google Scholar] [CrossRef]
- Pasqua, L.; De Napoli, I.E.; De Santo, M.; Greco, M.; Catizzone, E.; Lombardo, D.; Montera, G.; Comandè, A.; Nigro, A.; Morelli, C.; et al. Mesoporous Silica-Based Hybrid Materials for Bone-Specific Drug Delivery. Nanoscale Adv. 2019, 1, 3269–3278. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2/g) | Pore Volume (cm3/g) | Pores Mean Diameter (nm) |
|---|---|---|---|
| LPMS_100_2 | 834 | 0.49 | 10–12 |
| LPMS_140_2 | 634 | 0.60 | 14 |
| LPMS_220_2 | 536 | 0.70 | 16 |
| LPMS_100_24 | 626 | 0.93 | 23 |
| LPMS_140_24 | 428 | 1.09 | 17–23 |
| LPMS_200_24 | 348 | 0.96 | 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banche-Niclot, F.; Montalbano, G.; Fiorilli, S.; Vitale-Brovarone, C. PEG-Coated Large Mesoporous Silicas as Smart Platform for Protein Delivery and Their Use in a Collagen-Based Formulation for 3D Printing. Int. J. Mol. Sci. 2021, 22, 1718. https://doi.org/10.3390/ijms22041718
Banche-Niclot F, Montalbano G, Fiorilli S, Vitale-Brovarone C. PEG-Coated Large Mesoporous Silicas as Smart Platform for Protein Delivery and Their Use in a Collagen-Based Formulation for 3D Printing. International Journal of Molecular Sciences. 2021; 22(4):1718. https://doi.org/10.3390/ijms22041718
Chicago/Turabian StyleBanche-Niclot, Federica, Giorgia Montalbano, Sonia Fiorilli, and Chiara Vitale-Brovarone. 2021. "PEG-Coated Large Mesoporous Silicas as Smart Platform for Protein Delivery and Their Use in a Collagen-Based Formulation for 3D Printing" International Journal of Molecular Sciences 22, no. 4: 1718. https://doi.org/10.3390/ijms22041718
APA StyleBanche-Niclot, F., Montalbano, G., Fiorilli, S., & Vitale-Brovarone, C. (2021). PEG-Coated Large Mesoporous Silicas as Smart Platform for Protein Delivery and Their Use in a Collagen-Based Formulation for 3D Printing. International Journal of Molecular Sciences, 22(4), 1718. https://doi.org/10.3390/ijms22041718