
ATF3-Induced Mammary Tumors Exhibit Molecular Features of Human Basal-Like Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
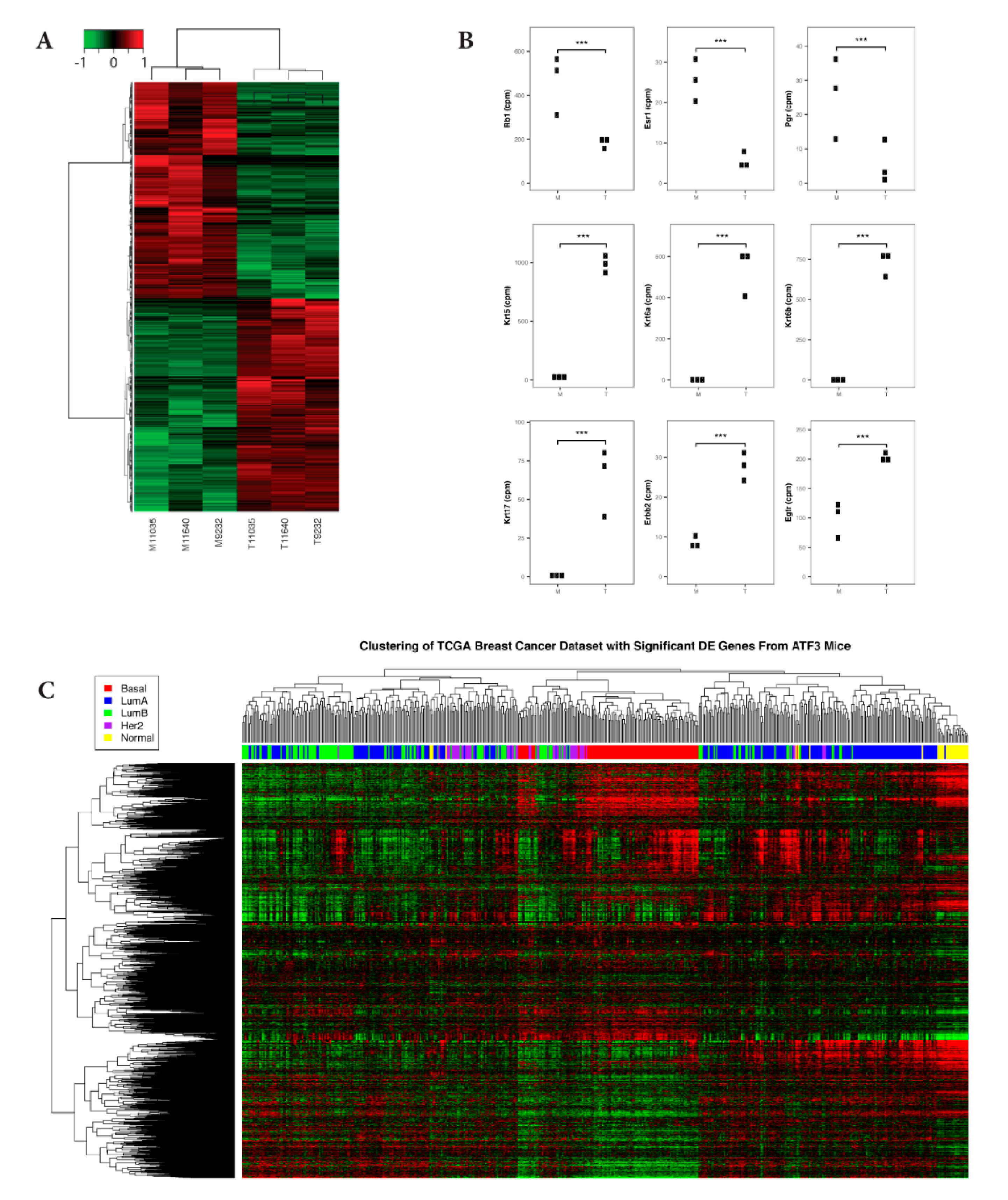
2.1. Gene Expression Pattern of ATF3-Induced Mammary Tumors is Consistent with Characteristics of Human Basal-Like Breast Cancers
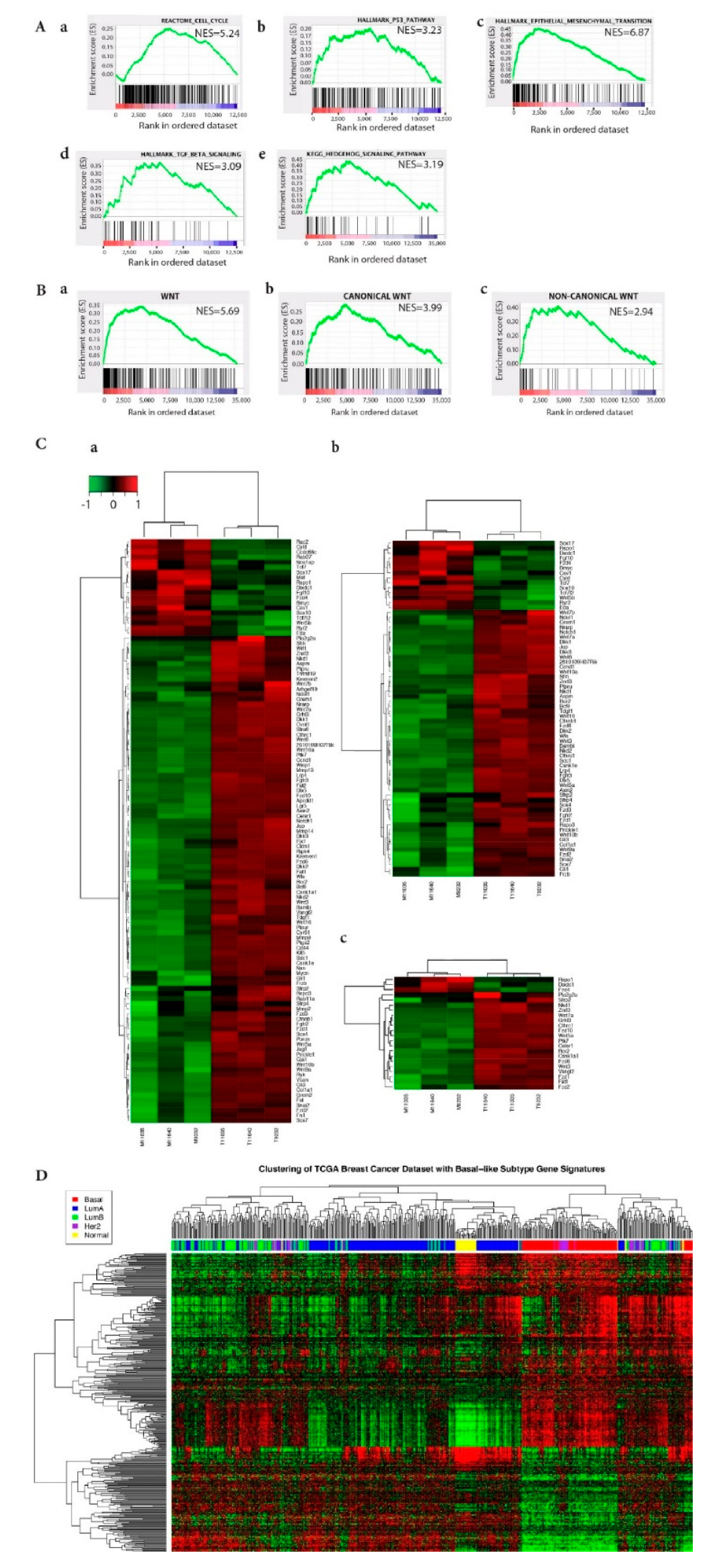
2.2. EMT, Wnt Signaling, and Stem Cell Biology are Implicated in ATF3-Induced Mammary Tumors
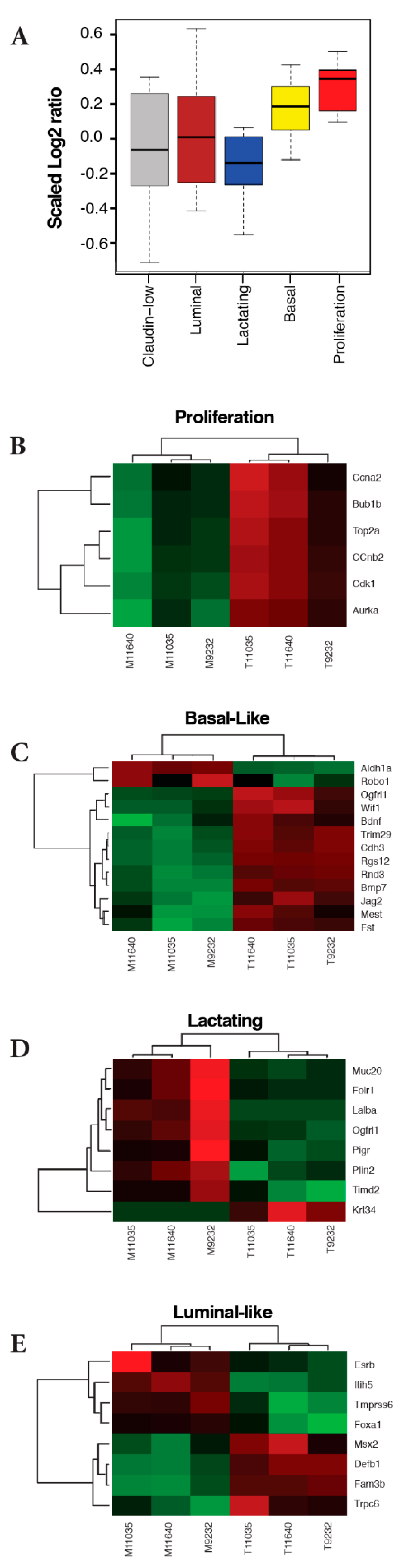
2.3. ATF3 Mammary Tumors Resemble Three Basal-Like Breast Cancer Mouse Models
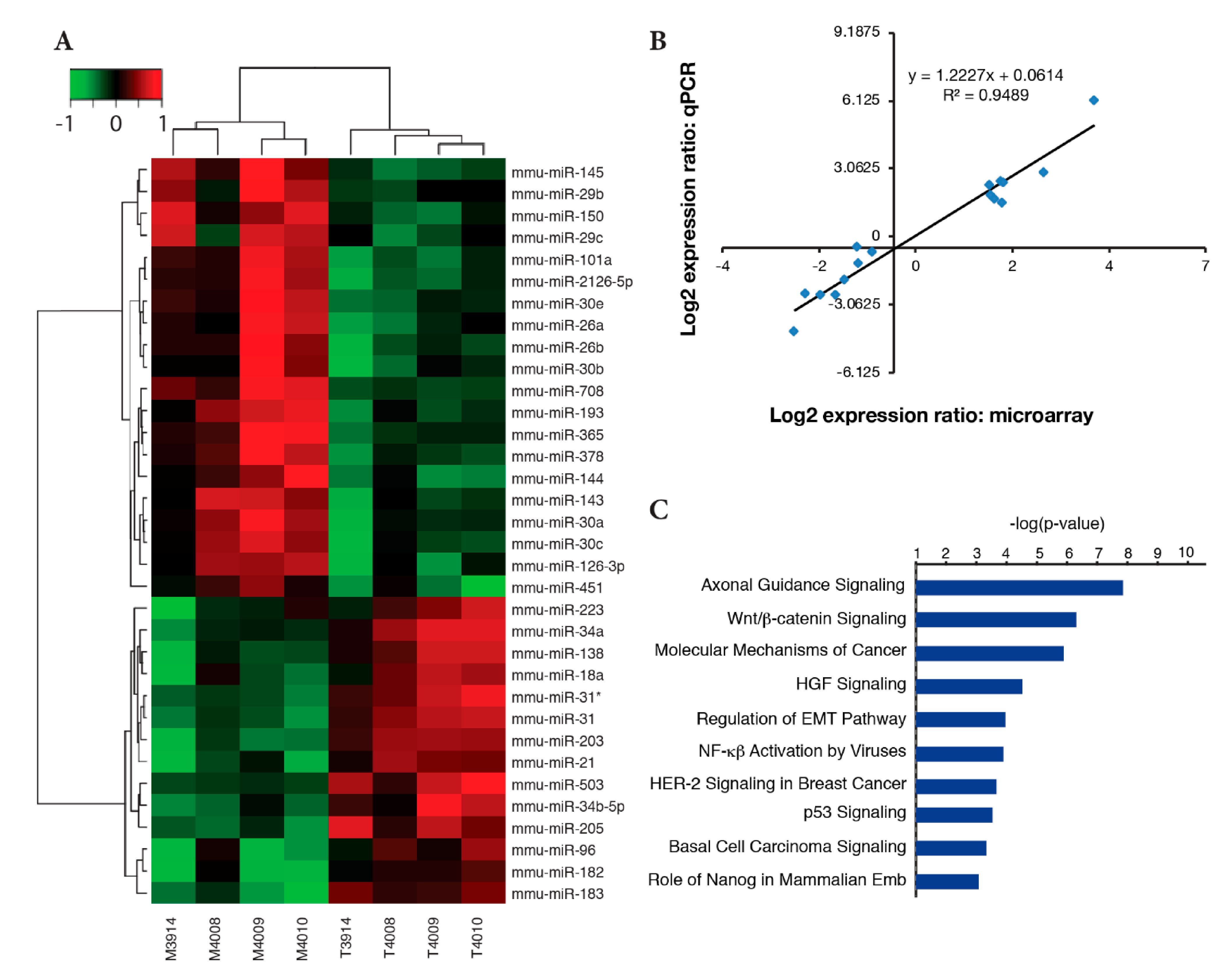
2.4. Identification of Differentially Expressed miRNAs
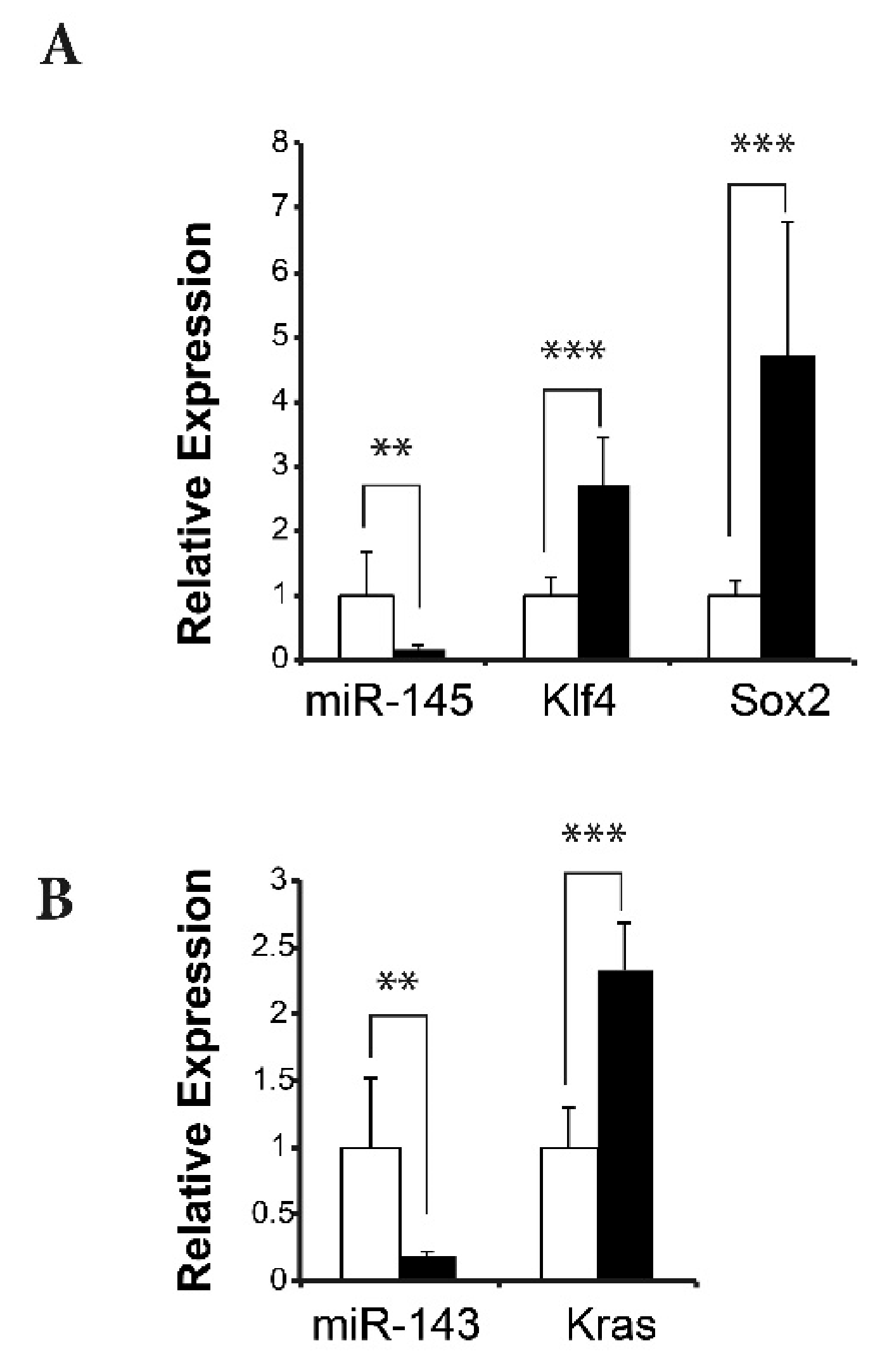
2.5. Reduced Mir145 and Mir143 Expression in ATF3 Tumors Upregulates Transcription Factors Klf4 and Sox2 and Cancer Stem Cell-Related Gene Kras
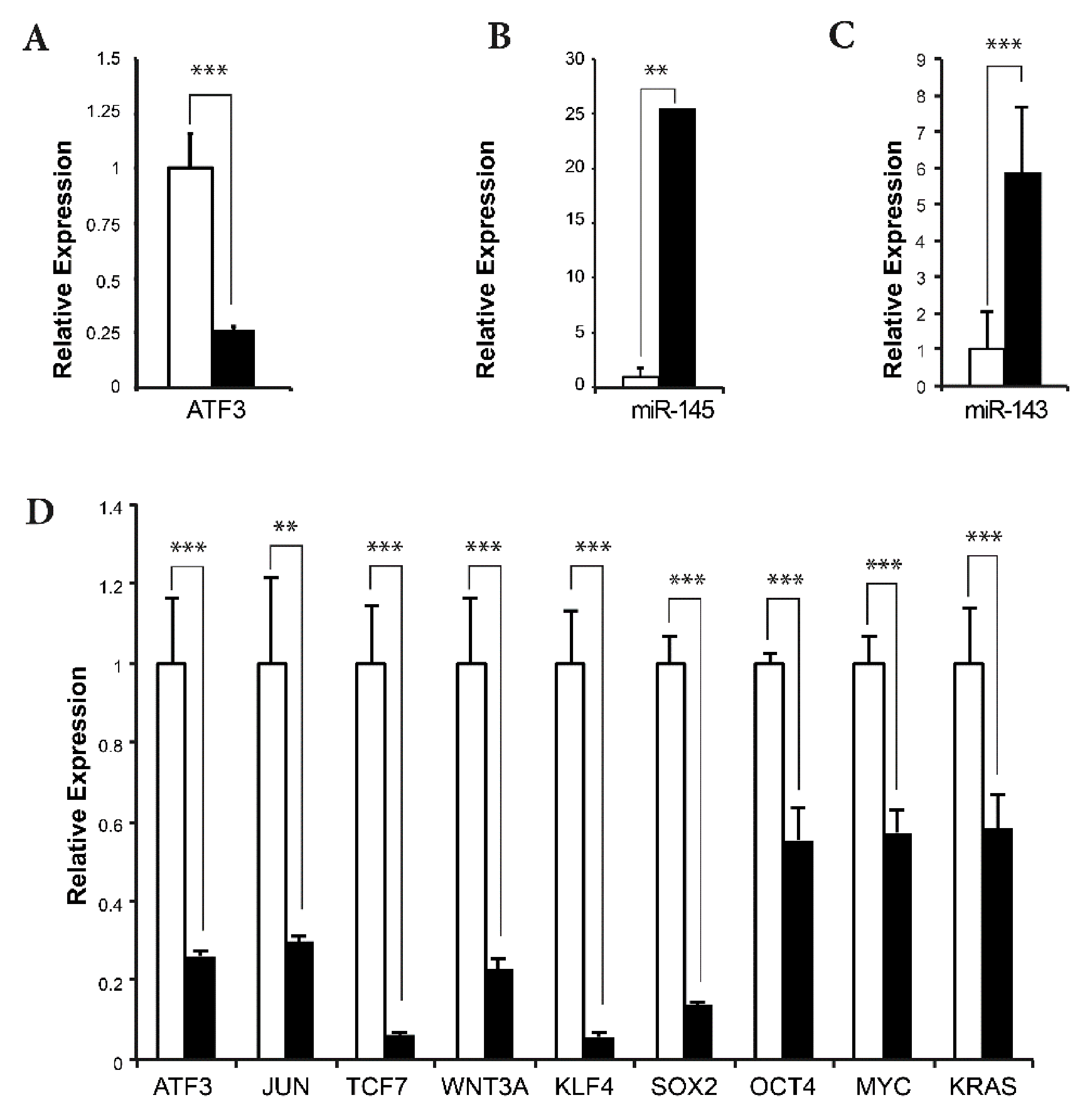
2.6. Effects of ATF3 Knockdown on the Expression of MIR145/143
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. miRNA Microarray and RT-qPCR Gene Expression Analysis
4.4. mRNA RNA-Seq Sequencing
4.5. Bioinformatics Analysis
4.5.1. MiRNA Microarray Analysis
4.5.2. RNA-Seq Data Analysis
4.5.3. Ingenuity Pathway Analysis (IPA) and the Integration of mRNA and miRNA Expression Data
4.5.4. Gene Expression Clustering and Gene Set Enrichment Analysis (GSEA)
4.5.5. PAM50 Correlation Test
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Badve, S.; Dabbs, S.J.; Schnitt, F.L.; Baehner, T.; Decker, V.; Eusebi, S.B.; Fox, S.; Ichihara, J.; Jacquemier, S.R.; Lakhani, J.; et al. Basal-like and triple-negative breast cancers, A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Arantes, L.; Yan, K.; Kiguchi, M.J.M.; Sahin, H.D.; Thames, C.M.A.; Macleod, M.C. The transcription factor ATF3 acts as an oncogene in mouse mammary tumorigenesis. BMC Cancer 2008, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Dewille, J.W.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Wolfgang, D.K.; Marsee, A.E.A.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. 1999, 7, 321–335. [Google Scholar] [PubMed]
- Amundson, S.A.; Bittner, Y.; Chen, J.; Trent, P.M.; Fornace, A. Fluorescent cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene 1999, 18, 3666–3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Gu, K.J.; Powell, H.; Mistry, P.; Simhambhatla, C.M.; Aldaz, S.G.; MacLeod, M.C. Response of human mammary epithelial cells to DNA damage induced by BPDE, Involvement of novel regulatory pathways. Carcinogenesis 2003, 24, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Ameri, K.; Hammond, E.M.; Culmseee, C.; Raida, M.; Katschinski, D.M.; Wenger, R.H.; Wagner, E.; Davis, R.J.; Hai, T.; Denko, N.; et al. Induction of activating transcription factor 3 by anoxia is independent of p53 and the hypoxic HIF signalling pathway. Oncogene 2007, 26, 284–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Wolford, C.C.; Chang, Y.S.; McConoughey, S.J.; Ramsey, S.A.; Aderem, A.; Hai, T. ATF3, an adaptive-response gene, enhances TGF{beta} signaling and cancer-initiating cell features in breast cancer cells. J. Cell. Sci. 2010, 123, 3558–3565. [Google Scholar] [CrossRef] [Green Version]
- Mathiasen, D.P.; Egebjerg, S.H.; Andersen, B.; Rafn, P.; Puustinen, A.; Khanna, M.; Daugaard, E.; Valo, S.; Tuomela, T.; Bottzauw, C.F.; et al. Identification of a c-Jun N-terminal kinase-2-dependent signal amplification cascade that regulates c-Myc levels in ras transformation. Oncogene 2012, 31, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Powell, J.; Shen, H.; Thames, C.M.A.; MacLeod, M.C. Activation of the canonical Wnt/beta-catenin pathway in ATF3-induced mammary tumors. PLoS ONE 2011, 6, e16515. [Google Scholar]
- Zhang, M.; Atkinson, R.L.; Rosen, J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3522–3527. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Samuelson, C.B.; Cui, Y.S.; Gerber, D.A. MAPK/ERK and Wnt/beta-Catenin pathways are synergistically involved in proliferation of Sca-1 positive hepatic progenitor cells. Biochem. Biophys Res. Commun. 2011, 409, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, C.; Guerrero, F.; Vicente, A.; Cordoba, M.; Esteller, S.; Ropero, F.G.; Lera, J.M. Differences and molecular immunohistochemical parameters in the subtypes of infiltrating ductal breast cancer. Am. J. Clin. Pathol. 2008, 130, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.; Foekens, J.A.; Martens, J.W. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [Green Version]
- DiMeo, T.A.; Anderson, P.; Phadke, C.; Fan, C.M.; Perou, S.N.; Kuperwasser, C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009, 69, 5364–5373. [Google Scholar] [CrossRef] [Green Version]
- Mayer, E.L.; Partridge, L.N.; Harris, R.S.; Gelman, S.T.; Schumer, H.J.B.; Winer, E.P. Tolerability of and adherence to combination oral therapy with gefitinib and capecitabine in metastatic breast cancer. Breast Cancer Res. Treat. 2009, 117, 615–623. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Das, L.M.; Heiser, S.; Bhattacharya, D.; Siwak, R.; Gendelman, N.; Bayani, N.J.; Wang, R.M.; Neve, Y.; Guan, Z.; et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 69, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Geyer, F.C.; Savage, M.; Arnedos, M.B.; Lambros, A.M.; Reis-Filho, J.S. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef]
- Teuliere, J.; Faraldo, M.M.; Deugnier, M.A.; Shtutman, M.; Ben-Ze'ev, A.; Thiery, J.P.; Glukhova, M.A. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development 2005, 132, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Kwok, S.; Rittling, N.C.; Partridge, C.S.; Benson, M.; Thiyagaraj, N.S.; Selvamurugan, N. Transforming growth factor-beta1 regulation of ATF-3 and identification of ATF-3 target genes in breast cancer cells. J. Cell Biochem. 2009, 108, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, A.D.; Herschkowitz, J.; Usary, J.C.; Harrell, B.T.; Spike, J.R.; Adams, M.I.T.; Brown, S.E.; Egan, G.M.; Wahl, J.M.R.; Perou, C.M. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.X.; Lin, H. The miRNA Regulation of Stem Cells. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 83–95. [Google Scholar]
- Micalizzi, D.S.; Christensen, P.; Jedlicka, R.D.; Coletta, A.E.; Baron, J.C.; Harrell, K.B.; Horwitz, D.; Billheimer, K.A.; Heichman, A.L.; Welm, W.P.S.; et al. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J. Clin. Investig. 2009, 119, 2678–2690. [Google Scholar] [CrossRef]
- Scimeca, M.; Antonacci, D.; Colombo, R.; Bonfiglio, O.C.B.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumour Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer, Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Zardawi, J.S.; O’Toole, S.A.; Sutherland, R.L.; Musgrove, E.A. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol. Histopathol. 2009, 24, 385–398. [Google Scholar]
- Ganapathy, V.; Ge, A.; Grazioli, W.; Xie, W.B.; Kang, S.; Lonning, J.M.; Yingling, S.; Biswas, G.R.M.; Reiss, M. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 2010, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guen, V.J.; Chavarria, C.; Kroger, X.; Ye, R.A.W.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roarty, K.; Pfefferle, C.J.; Creighton, C.M.P.; Rosen, J.M. Ror2-mediated alternative Wnt signaling regulates cell fate and adhesion during mammary tumor progression. Oncogene 2017, 36, 5958–5968. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, Y.; Zhang, W.; Zhou, T.; Zhu, Q.; Ruan, H.; Chen, J.; Fang, F.; Zhou, J.S.; Yang, X. WNT5B governs the phenotype of basal-like breast cancer by activating WNT signaling. Cell Commun. Signal. 2019, 17, 109. [Google Scholar] [CrossRef] [Green Version]
- Cordes, K.R.; Sheehy, M.P.; White, E.C.; Berry, S.U.; Morton, A.N.; Muth, T.H.; Lee, J.M.; Miano, K.N.I.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Iorio, M.V.; Ferracin, C.G.; Liu, A.; Veronese, R.; Spizzo, S.; Sabbioni, E.; Magri, M.; Pedriali, M.; Fabbri, M.; Campiglio, S.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [Green Version]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Sempere, L.F.; Christensen, A.; Silahtaroglu, M.; Bak, C.V.; Heath, G.; Schwartz, W.; Wells, S.K.; Cole, C.N. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007, 67, 11612–11620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockmeyer, C.L.; Christgen, M.; Muller, S.; Fischer, P.; Ahrens, F.; Langer, H.K.; Lehmann, U. MicroRNA profiles of healthy basal and luminal mammary epithelial cells are distinct and reflected in different breast cancer subtypes. Breast Cancer Res. Treat 2011, 130, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Radojicic, J.; Zaravinos, T.; Vrekoussis, M.; Kafousi, D.A.S.; Stathopoulos, E.N. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle 2011, 10, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.; Zhu, F.; Wu, H.; Wu, V.; Walia, S.; Kumar, R.; Elble, K.W.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Papagiannakopoulos, G.; Pan, J.A.T.; Kosik, K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Kent, O.A.; Chivukula, M.; Mullendore, E.A.; Wentzel, G.; Feldmann, K.H.; Lee, S.; Liu, S.D.; Leach, A.M.; Mendell, J.T. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010, 24, 2754–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, K.; Link, K.; Friedrich, A.; Petzold, A.; Niedostatek, O.; Schoffer, C.; Werner, S.J.; Klug, A.; Werner, A.; Gatzweiler, B.; et al. Impact of breast cancer subtypes and patterns of metastasis on outcome. Breast Cancer Res. Treat. 2015, 150, 621–629. [Google Scholar] [CrossRef]
- Bhargava, R.; Beriwal, K.M.; Dabbs, D.J. CK5 is more sensitive than CK5/6 in identifying the basal-like phenotype of breast carcinoma. Am. J. Clin. Pathol. 2008, 130, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Alshareeda, A.T.; Soria, D.; Garibaldi, J.M.; Rakha, E.; Nolan, C.; Ellis, I.O.; Green, A.R. Characteristics of basal cytokeratin expression in breast cancer. Breast Cancer Res. Treat 2013, 139, 23–37. [Google Scholar] [CrossRef]
- Wang, A.; Arantes, C.; Conti, M.M.; Macleod, M.C. Epidermal hyperplasia and oral carcinoma in mice overexpressing the transcription factor ATF3 in basal epithelial cells. Mol. Carcinog. 2007, 46, 476–487. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: http://www.Rproject.org/ (accessed on 15 February 2021).
- Ritchie, M.E.; Phipson, D.; Wu, Y.; Hu, C.W.; Law, W.S.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Huber, W.; Sultmann, A.P.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Phipson, B.; Lee, I.J.; Majewski, W.S.A.; Smyth, G.K. Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Benjamnini, Y.; Hochberg, Y. Controlling the False Discovery Rate, A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Kim, D.; Pertea, C.; Trapnell, H.; Pimentel, R.K.; Salzberg, S.L. TopHat2, Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Tatusova, G.R.B.; Maglott, D.R. NCBI Reference Sequences (RefSeq), Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.M.; McCarthy, D.J.; Smyth, G.K. edgeR, A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic. Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, L.; Gaddis, S.; Coletta, L.D.; Repass, J.; Powell, K.L.; Simper, M.S.; Chen, Y.; Byrom, M.; Zhong, Y.; Lin, K.; et al. ATF3-Induced Mammary Tumors Exhibit Molecular Features of Human Basal-Like Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2353. https://doi.org/10.3390/ijms22052353
Yan L, Gaddis S, Coletta LD, Repass J, Powell KL, Simper MS, Chen Y, Byrom M, Zhong Y, Lin K, et al. ATF3-Induced Mammary Tumors Exhibit Molecular Features of Human Basal-Like Breast Cancer. International Journal of Molecular Sciences. 2021; 22(5):2353. https://doi.org/10.3390/ijms22052353
Chicago/Turabian StyleYan, Leqin, Sally Gaddis, Luis Della Coletta, John Repass, Katherine Leslie Powell, Melissa S. Simper, Yueping Chen, Michelle Byrom, Yi Zhong, Kevin Lin, and et al. 2021. "ATF3-Induced Mammary Tumors Exhibit Molecular Features of Human Basal-Like Breast Cancer" International Journal of Molecular Sciences 22, no. 5: 2353. https://doi.org/10.3390/ijms22052353
APA StyleYan, L., Gaddis, S., Coletta, L. D., Repass, J., Powell, K. L., Simper, M. S., Chen, Y., Byrom, M., Zhong, Y., Lin, K., Liu, B., Lu, Y., Shen, J., & MacLeod, M. C. (2021). ATF3-Induced Mammary Tumors Exhibit Molecular Features of Human Basal-Like Breast Cancer. International Journal of Molecular Sciences, 22(5), 2353. https://doi.org/10.3390/ijms22052353