
Toward the Pathogenicity of the SLC26A4 p.C565Y Variant Using a Genetically Driven Mouse Model

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
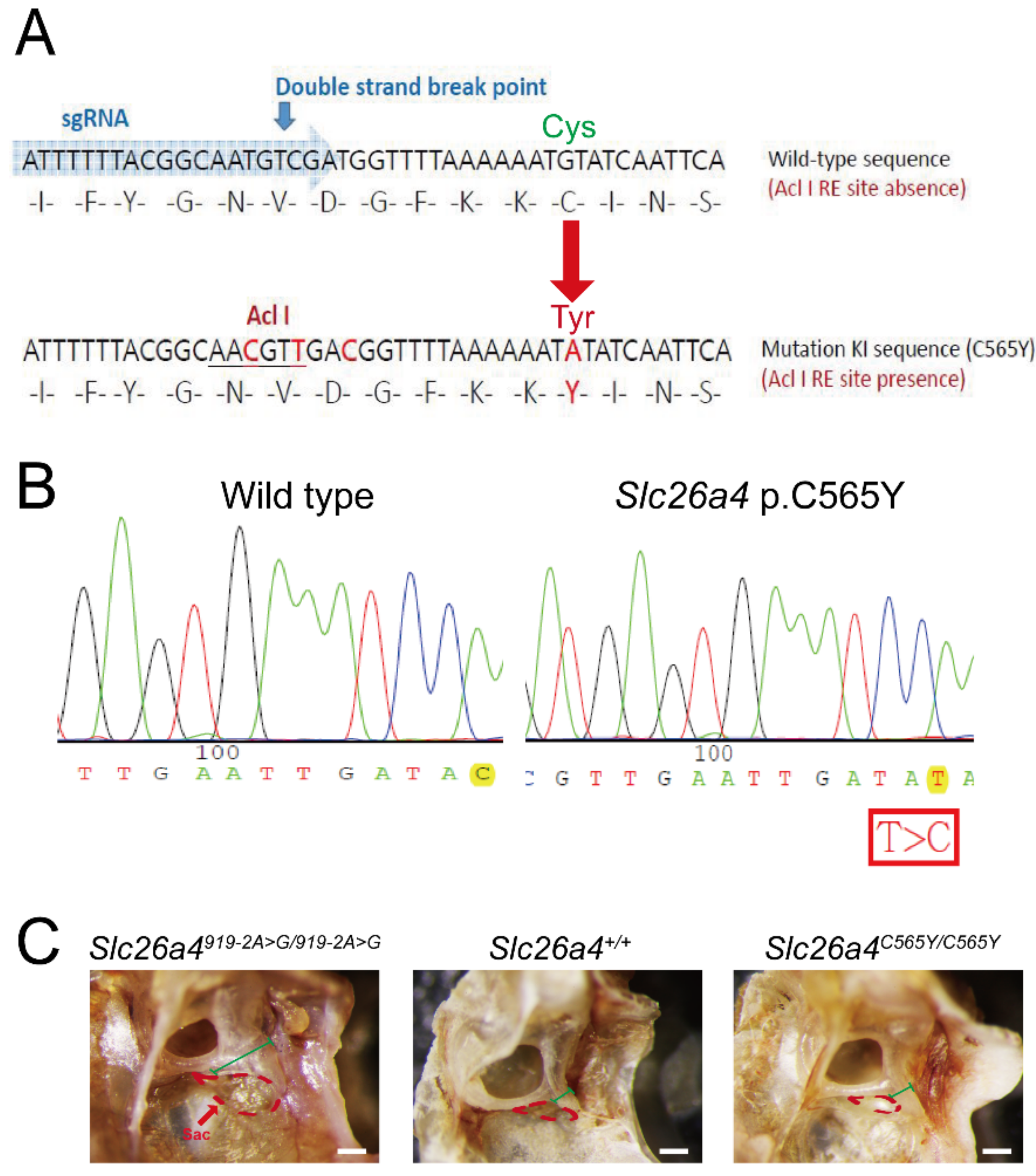
2.1. Gross Inner-Ear Morphology
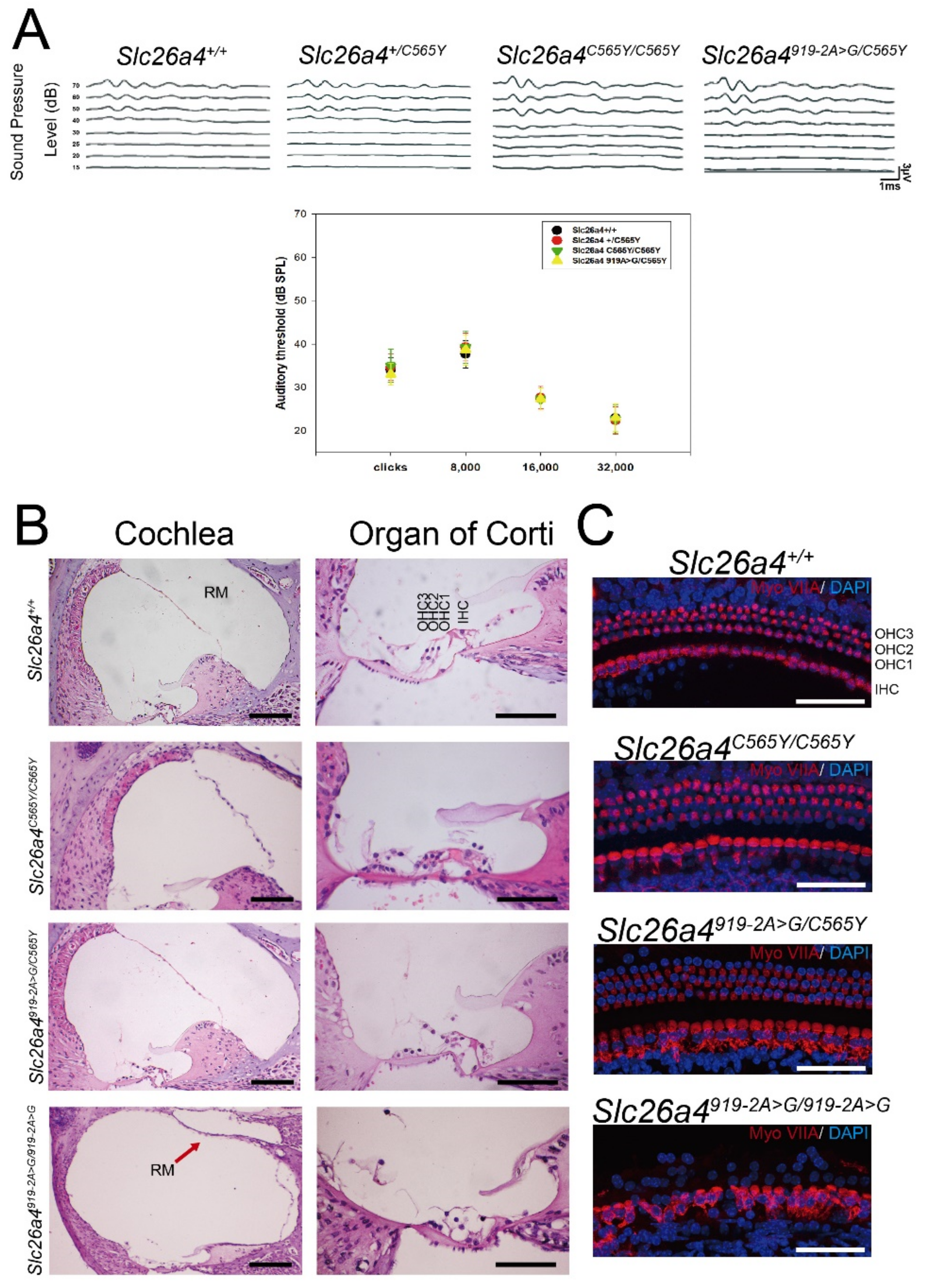
2.2. Auditory-Phenotype Evaluation and Cochlear Histology
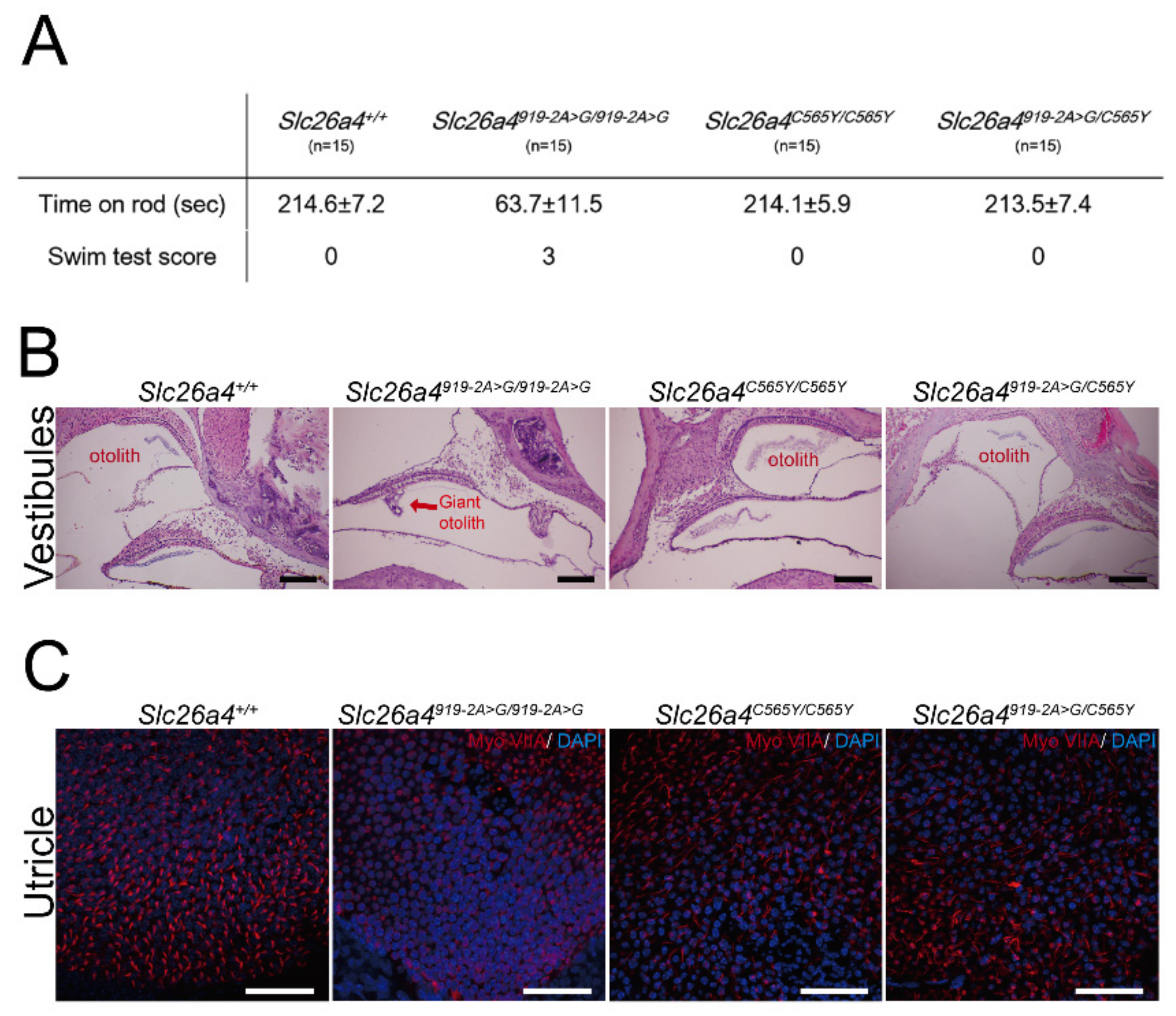
2.3. Vestibular-Function Evaluation and Histology of Vestibular Organs
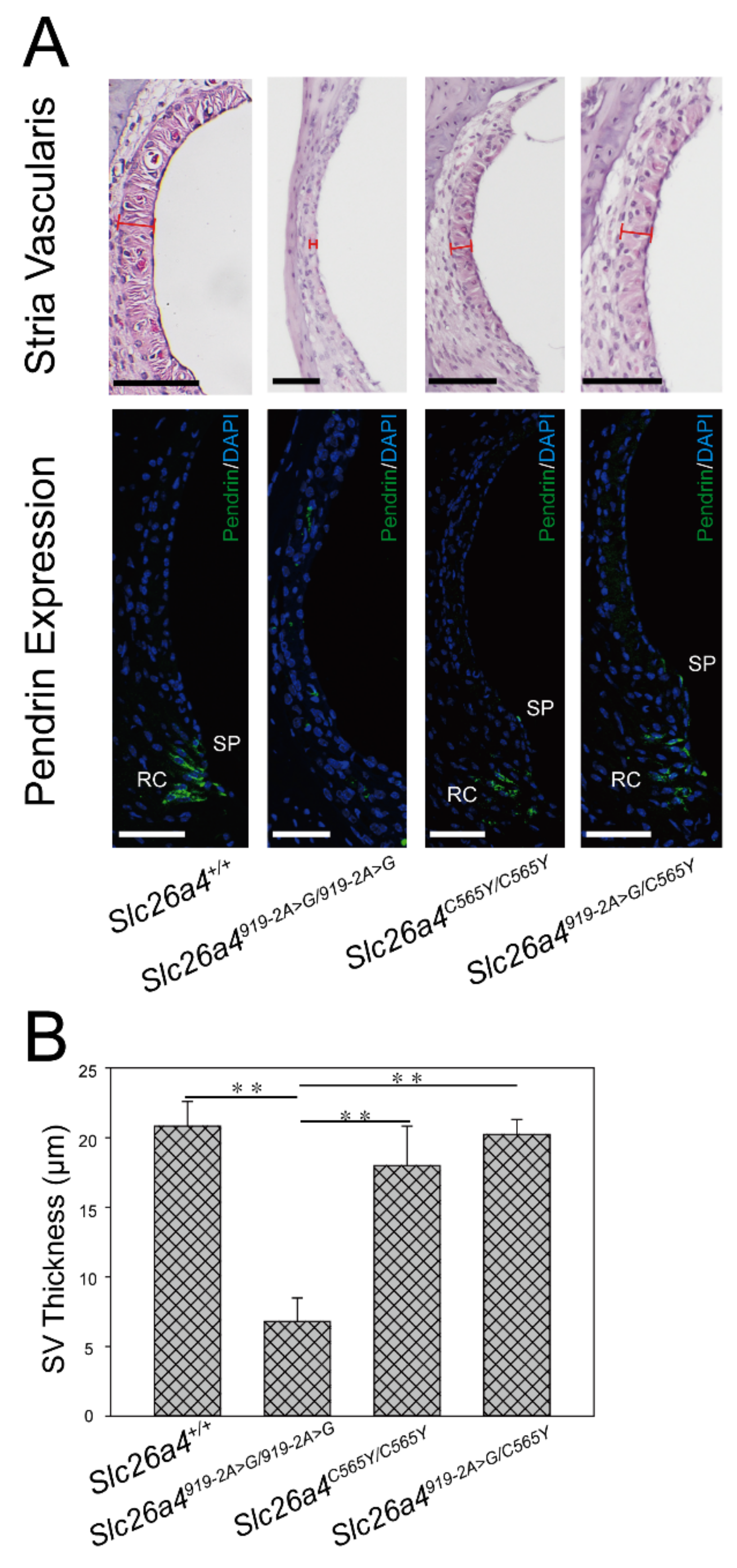
2.4. Morphology of Stria Vascularis
3. Discussion
4. Materials and Methods
4.1. Generation of Slc26a4C565Y/C565Y Knock-In Mice
4.2. Auditory Evaluations
4.3. Vestibular Evaluations
4.4. Inner-Ear Histology Studies
4.5. Pendrin Expression
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wolf, A.; Frohne, A.; Allen, M.; Parzefall, T.; Koenighofer, M.; Schreiner, M.M.; Schoefer, C.; Frei, K.; Lucas, T. A Novel Mutation in SLC26A4 Causes Nonsyndromic Autosomal Recessive Hearing Impairment. Otol. Neurotol. 2017, 38, 173–179. [Google Scholar] [CrossRef]
- Han, M.; Li, Z.; Wang, W.; Huang, S.; Lu, Y.; Gao, Z.; Wang, L.; Kang, N.; Li, L.; Liu, Y.; et al. A quantitative cSMART assay for noninvasive prenatal screening of autosomal recessive nonsyndromic hearing loss caused by GJB2 and SLC26A4 mutations. Genet. Med. 2017, 19, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Everett, L.A.; Morsli, H.; Wu, D.K.; Green, E.D. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 1999, 96, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Pelzl, L.; Pakladok, T.; Pathare, G.; Fakhri, H.; Michael, D.; Wagner, C.A.; Paulmichl, M.; Lang, F. DOCA Sensitive Pendrin Expression in Kidney, Heart, Lung and Thyroid Tissues. Cell. Physiol. Biochem. 2012, 30, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Everett, L.A.; Lalwani, A.K.; Desmukh, D.; Friedman, T.B.; Green, E.D.; Wilcox, E.R. A mutation in PDS causes non-syndromic recessive deafness. Nat. Genet. 1998, 18, 215–217. [Google Scholar] [CrossRef]
- Mori, T.; Westerberg, B.D.; Atashband, S.; Kozak, F.K. Natural history of hearing loss in children with enlarged vestibular aqueduct syndrome. J. Otolaryngol. Head Neck Surg. 2008, 37, 112–118. [Google Scholar] [PubMed]
- Yoon, J.S.; Park, H.-J.; Yoo, S.-Y.; Namkung, W.; Jo, M.J.; Koo, S.K.; Lee, W.-S.; Kim, K.H.; Lee, M.G. Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 2008, 45, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Okuyama, S.; Kumano, S.; Iida, K.; Hamana, H.; Murakoshi, M.; Kobayashi, T.; Usami, S.; Ikeda, K.; Haga, Y.; et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear. Res. 2010, 270, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Stewart, A.K.; Madeo, A.C.; Pryor, S.P.; Lenhard, S.; Kittles, R.; Eisenman, D.; Kim, H.J.; Niparko, J.; Thomsen, J.; et al. Hypo-FunctionalSLC26A4variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: Genotype-phenotype correlation or coincidental polymorphisms? Hum. Mutat. 2009, 30, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, E.W.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, A.J.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Van Hauwe, P.; Everett, L.A.; Coucke, P.; Scott, D.A.; Kraft, M.L.; Ris-Stalpers, C.; Bolder, C.; Otten, B.; De Vijlder, J.J.M.; Dietrich, N.L.; et al. Two Frequent Missense Mutations in Pendred Syndrome. Hum. Mol. Genet. 1998, 7, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S.-I. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef] [Green Version]
- Everett, L.A.; Belyantseva, I.A.; Noben-Trauth, K.; Cantos, R.; Chen, A.; Thakkar, S.I.; Hoogstraten-Miller, S.L.; Kachar, B.; Wu, D.K.; Green, E.D. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Mol. Genet. 2001, 10, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Dror, A.A.; Politi, Y.; Shahin, H.; Lenz, D.R.; Dossena, S.; Nofziger, C.; Fuchs, H.; De Angelis, M.H.; Paulmichl, M.; Weiner, S.; et al. Calcium Oxalate Stone Formation in the Inner Ear as a Result of an Slc26a4 Mutation. J. Biol. Chem. 2010, 285, 21724–21735. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-C.; Wu, C.-C.; Shen, W.-S.; Yang, T.-H.; Yeh, T.-H.; Chen, P.-J.; Yu, I.-S.; Lin, S.-W.; Wong, J.-M.; Chang, Q.; et al. Establishment of a Knock-In Mouse Model with the SLC26A4 c.919-2A>G Mutation and Characterization of Its Pathology. PLoS ONE 2011, 6, e22150. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Wu, C.-C.; Yang, T.-H.; Lin, Y.-H.; Yu, I.-S.; Lin, S.-W.; Chang, Q.; Lin, X.; Wong, J.-M.; Hsu, C.-J. Differences in the Pathogenicity of the p.H723R Mutation of the Common Deafness-Associated SLC26A4 Gene in Humans and Mice. PLoS ONE 2013, 8, e64906. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Zhu, H.; Li, Z.; Zhang, S.; Zhang, A.; Zhang, T.; Fu, X.; Sun, D.; Zhang, J.; Gao, J. A knock-in mouse model of Pendred syndrome with Slc26a4 L236P mutation. Biochem. Biophys. Res. Commun. 2019, 515, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, H.-M.; Ito, T.; Lee, K.-Y.; Li, X.; Monahan, K.; Wen, Y.; Wilson, E.; Kurima, K.; Saunders, T.L.; et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J. Clin. Investig. 2011, 121, 4516–4525. [Google Scholar] [CrossRef]
- Choi, H.J.; Lee, H.J.; Choi, J.Y.; Jeon, I.H.; Noh, B.; Devkota, S.; Lee, H.-W.; Eo, S.K.; Choi, J.Y.; Lee, M.G.; et al. DNAJC14 Ameliorates Inner Ear Degeneration in the DFNB4 Mouse Model. Mol. Ther. Methods Clin. Dev. 2020, 17, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, H.; Miwa, T.; Kim, M.Y.; Choi, B.Y.; Orita, Y.; Minoda, R. Prenatal electroporation-mediated gene transfer restores Slc26a4 knock-out mouse hearing and vestibular function. Sci. Rep. 2019, 9, 17979. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-M.; Wangemann, P. Epithelial Cell Stretching and Luminal Acidification Lead to a Retarded Development of Stria Vascularis and Deafness in Mice Lacking Pendrin. PLoS ONE 2011, 6, e17949. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Li, X.; Kurima, K.; Choi, B.Y.; Wangemann, P.; Griffith, A.J. Slc26a4-insufficiency causes fluctuating hearing loss and stria vascularis dysfunction. Neurobiol. Dis. 2014, 66, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royaux, I.E.; Belyantseva, I.A.; Wu, T.; Kachar, B.; Everett, L.A.; Marcus, D.C.; Green, E.D. Localization and Functional Studies of Pendrin in the Mouse Inner Ear Provide Insight About the Etiology of Deafness in Pendred Syndrome. J. Assoc. Res. Otolaryngol. 2003, 4, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangemann, P.; Itza, E.M.; Albrecht, B.; Wu, T.; Jabba, S.V.; Maganti, R.J.; Lee, J.H.; Everett, A.L.; Wall, S.M.; Royaux, E.I.; et al. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004, 2, 30. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-J.; Lee, S.-T.; Lee, M.-K.; Ji, Y.; Kim, J.-W.; Ki, C.-S. Estimation of carrier frequencies of six autosomal-recessive Mendelian disorders in the Korean population. J. Hum. Genet. 2012, 57, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.-Y.; Hattori, M.; Usami, S.-I. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, andPDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef]
- Suzuki, H.; Oshima, A.; Tsukamoto, K.; Abe, S.; Kumakawa, K.; Nagai, K.; Satoh, H.; Kanda, Y.; Iwasaki, S.; Usami, S.-I. Clinical characteristics and genotype–phenotype correlation of hearing loss patients withSLC26A4mutations. Acta Otolaryngol. 2007, 127, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Yang, T.; Prasad, S.; Sorensen, J.L.; Nishimura, C.J.; Kimberling, W.J.; Smith, R.J.H. Genotype–phenotype correlations for SLC26A4-related deafness. Qual. Life Res. 2007, 122, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Chen, P.-J.; Hsu, C.-J. Specificity of SLC26A4 Mutations in the Pathogenesis of Inner Ear Malformations. Audiol. Neurotol. 2005, 10, 234–242. [Google Scholar] [CrossRef]
- Miyagawa, M.; the Deafness Gene Study Consortium; Nishio, S.-Y.; Usami, S.-I. Mutation spectrum and genotype–phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Lu, Y.-C.; Chen, P.-J.; Yeh, P.-L.; Su, Y.-N.; Hwu, W.-L.; Hsu, C.-J. Phenotypic Analyses and Mutation Screening of the SLC26A4 and FOXI1 Genes in 101 Taiwanese Families with Bilateral Nonsyndromic Enlarged Vestibular Aqueduct (DFNB4) or Pendred Syndrome. Audiol. Neurotol. 2009, 15, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jung, J.; Shin, J.; Song, M.; Kim, S.; Lee, J.-H.; Lee, K.-A.; Shin, S.; Kim, U.-K.; Bok, J.; et al. Correlation between genotype and phenotype in patients with bi-allelicSLC26A4mutations. Clin. Genet. 2013, 86, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Rah, Y.C.; Kim, A.R.; Koo, J.-W.; Lee, J.H.; Oh, S.-H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to theSLC26A4genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.R.; Chattaraj, P.; Munjal, T.; Honda, K.; King, K.A.; Zalewski, C.K.; Chien, W.W.; Brewer, C.C.; Griffith, A.J. SLC26A4-linked CEVA haplotype correlates with phenotype in patients with enlargement of the vestibular aqueduct. BMC Med. Genet. 2019, 20, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Sone, M.; Naganawa, S.; Nakashima, T. Patient with an SLC26A4 gene mutation who had low-frequency sensorineural hearing loss and endolymphatic hydrops. J. Laryngol. Otol. 2015, 129, 95–97. [Google Scholar] [CrossRef]
- Zhang, B.-Y.; Zhang, J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 6987–6992. [Google Scholar]
- Li, X.; Sanneman, J.D.; Harbidge, D.G.; Zhou, F.; Ito, T.; Nelson, R.; Picard, N.; Chambrey, R.; Eladari, D.; Miesner, T.; et al. SLC26A4 Targeted to the Endolymphatic Sac Rescues Hearing and Balance in Slc26a4 Mutant Mice. PLoS Genet. 2013, 9, e1003641. [Google Scholar] [CrossRef]
- Hardisty-Hughes, E.R.; Parker, A.; Brown, S.D.M. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat. Protoc. 2010, 5, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slc26a4−/− [13] | Slc26a4loop/loop [14] | Tg[E];Tg[R];Slc26a4Δ/Δ [18] | Slc26a4919A>G/919A>G [15] | Slc26a4H723R/H723R [16] | Slc26a4 L236P [17] | hH723R Tg [19] | Slc26a4C565Y/C565Y | |
|---|---|---|---|---|---|---|---|---|
| Audiological phenotypes | Profound hearing loss (>100 dB SPL) | Profound hearing loss (>100 dB SPL) | Hearing levels depend on the time of Slc26a4 expression. Doxycycline initiation at E18.5 (IE18.5) results in partial hearing loss | Profound hearing loss (>120 dB SPL) | Normal | Moderate-to-profound hearing loss in mice at 1 month. No progressive hearing loss up to 9 months | Profound hearing loss (>100 dB SPL) | Normal |
| Cochlear hair cells | Severe degeneration of inner and outer hair cells by P45 | ND | Functionally intact at P25 to P35 | Severe degeneration of inner and outer hair cells at 6 w | Normal up to P60 | Different degrees of hair-cell degeneration and abnormal structures of stereocilia | Mild-to-severe degeneration of hair cells | Normal up to P90 |
| Stria vascularis | Atrophic | ND | No significant difference between wild-type, IE18.5, and discontinued at E17.5 (DE17.5) | Atrophic | Normal | Atrophic | Atrophic | Normal |
| Vestibular aqueduct and enndolymphatic hydrops | Enlarged | ND | Size depends on time of Slc26a4 expression. Significantly enlarged in E18.5 mice | Enlarged | Normal | ND | Enlarged | Normal |
| Vestibular phenotypes | Vestibular deficits, including head tilting, head bobbing, and circling | Variable vestibular deficits, including unsteady gait, circling and tilted body | ND | 46% of mice with head tilting and circling | Normal | 9 of 31 L236P mice had balance dysfunction. Vestibular dysfunction variable in L236P mice | ND | Normal |
| Vestibular hair cells | Severe degeneration of vestibular hair cells by P45 | Normal morphology of vestibular hair cells at 2 m | ND | Loss and degeneration of utricular hair cells | Normal | Normal | ND | Normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.-J.; Lu, Y.-C.; Yang, T.-H.; Chan, Y.-H.; Tsai, C.-Y.; Yu, I.-S.; Lin, S.-W.; Liu, T.-C.; Cheng, Y.-F.; Wu, C.-C.; et al. Toward the Pathogenicity of the SLC26A4 p.C565Y Variant Using a Genetically Driven Mouse Model. Int. J. Mol. Sci. 2021, 22, 2789. https://doi.org/10.3390/ijms22062789
Hu C-J, Lu Y-C, Yang T-H, Chan Y-H, Tsai C-Y, Yu I-S, Lin S-W, Liu T-C, Cheng Y-F, Wu C-C, et al. Toward the Pathogenicity of the SLC26A4 p.C565Y Variant Using a Genetically Driven Mouse Model. International Journal of Molecular Sciences. 2021; 22(6):2789. https://doi.org/10.3390/ijms22062789
Chicago/Turabian StyleHu, Chin-Ju, Ying-Chang Lu, Ting-Hua Yang, Yen-Hui Chan, Cheng-Yu Tsai, I-Shing Yu, Shu-Wha Lin, Tien-Chen Liu, Yen-Fu Cheng, Chen-Chi Wu, and et al. 2021. "Toward the Pathogenicity of the SLC26A4 p.C565Y Variant Using a Genetically Driven Mouse Model" International Journal of Molecular Sciences 22, no. 6: 2789. https://doi.org/10.3390/ijms22062789
APA StyleHu, C. -J., Lu, Y. -C., Yang, T. -H., Chan, Y. -H., Tsai, C. -Y., Yu, I. -S., Lin, S. -W., Liu, T. -C., Cheng, Y. -F., Wu, C. -C., & Hsu, C. -J. (2021). Toward the Pathogenicity of the SLC26A4 p.C565Y Variant Using a Genetically Driven Mouse Model. International Journal of Molecular Sciences, 22(6), 2789. https://doi.org/10.3390/ijms22062789