
Cardiac Protective Effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 Cells through Nrf2 Signaling via PI3K/AKT Pathways


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. KRL Dose-Dependently Inhibits DOX-Induced Cell Death
2.2. KRL Inhibits Natriuretic Peptide System
2.3. KRL Attenuates Activation of Matrix Metalloproteinases 2 and 9
2.4. Effect of KRL on DOX-Induced H9c2 Cytoskeletal Alterations
2.5. Effect of KRL on DOX-Induced Cell Survival Signaling
2.6. KRL Inhibits Phosphorylation of MAPKs in DOX-Induced Cardiotoxicity
2.7. KRL Suppresses Oxidative Stress in DOX-Induced Cardiomyocytes
2.8. KRL Enhances the Expression of Nrf2 in DOX-Treated H9c2 Cells
2.9. KRL Upregulates the Translocation of Nrf2
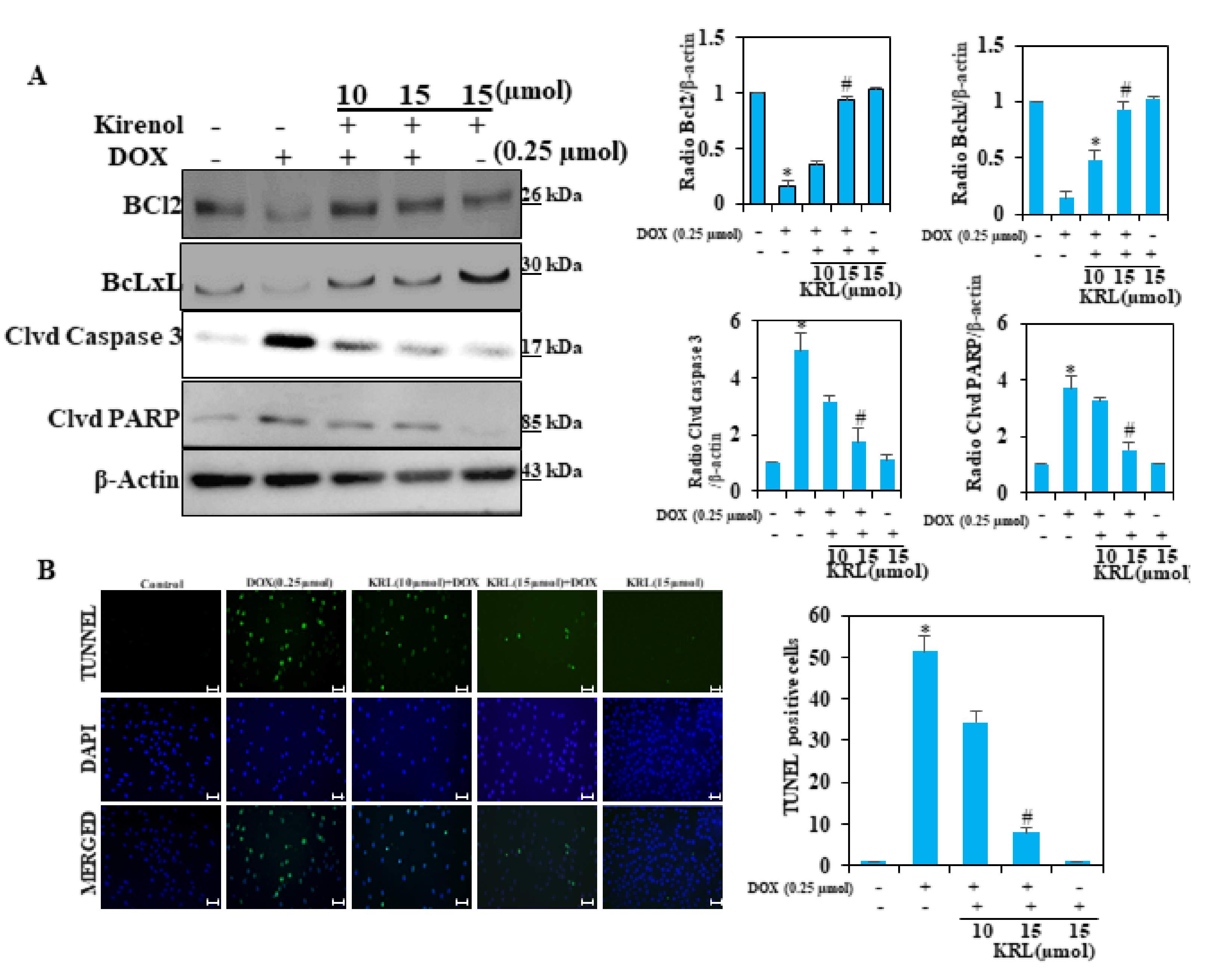
2.10. KRL Protects H9c2 Cells Against DOX-Induced Cardiomyocyte Apoptosis
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability
4.4. Preparation of Cytoplasmic and Nuclear Extracts
4.5. Western Blotting
4.6. Phalloidin Staining
4.7. Immunostaining
4.8. TUNEL Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kim, Y.A.; Cho, H.; Lee, N.; Jung, S.Y.; Sim, S.H.; Park, I.H.; Lee, S.; Lee, E.S.; Kim, H.J. Doxorubicin-induced heart failure in cancer patients: A cohort study based on the Korean National Health Insurance Database. Cancer Med. 2018, 7, 6084–6092. [Google Scholar] [CrossRef]
- Szmit, S.; Jurczak, W.; Zaucha, J.M.; Drozd-Sokołowska, J.; Spychałowicz, W.; Joks, M.; Długosz-Danecka, M.; Torbicki, A. Pre-existing arterial hypertension as a risk factor for early left ventricular systolic dysfunction following (R)-CHOP chemotherapy in patients with lymphoma. J. Am. Soc. Hypertens. 2014, 8, 791–799. [Google Scholar] [CrossRef]
- Vaitiekus, D.; Muckiene, G.; Vaitiekiene, A.; Maciuliene, D.; Vaiciuliene, D.; Ambrazeviciute, G.; Sereikaite, L.; Verikas, D.; Jurkevicius, R.; Juozaityte, E. Impact of arterial hypertension on doxorubicin-based chemotherapy-induced subclinical cardiac damage in breast cancer patients. Cardiovasc. Toxicol. 2020, 20, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Hou, C.W.; Shibu, M.A.; Day, C.H.; Pai, P.; Liu, Z.R.; Lin, T.Y.; Viswanadha, V.P.; Kuo, C.H.; Huang, C.Y. Protective effect of Co-enzyme Q10 On doxorubicin-induced cardiomyopathy of rat hearts. Environ. Toxicol. 2017, 32, 679–689. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Li, D.L.; Hill, J.A. Cardiomyocyte autophagy and cancer chemotherapy. J. Mol. Cell. Cardiol. 2014, 71, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Chen, Q.; Sun, Y.P.; Wang, X.; Lv, L.; Zhang, L.P.; Liu, J.S.; Zhao, S.; Wang, X.L. Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovasc. Ther. 2017, 35, e12277. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and-independent pathways. Cardiovasc. Res. 2013, 97, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daffu, G.; Del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [Green Version]
- Imam, F.; Al-Harbi, N.O.; Al-Harbi, M.M.; Ansari, M.A.; Al-Asmari, A.F.; Ansari, M.N.; Al-Anazi, W.A.; Bahashwan, S.; Almutairi, M.M.; Alshammari, M. Apremilast prevent doxorubicin-induced apoptosis and inflammation in heart through inhibition of oxidative stress mediated activation of NF-κB signaling pathways. Pharmacol. Rep. 2018, 70, 993–1000. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kuo, W.-W.; Lo, J.-F.; Ho, T.-J.; Pai, P.-y.; Chiang, S.-F.; Chen, P.-Y.; Tsai, F.-J.; Tsai, C.-H. Doxorubicin attenuates CHIP-guarded HSF1 nuclear translocation and protein stability to trigger IGF-IIR-dependent cardiomyocyte death. Cell Death Dis. 2016, 7, e2455. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Arola, O.J.; Saraste, A.; Pulkki, K.; Kallajoki, M.; Parvinen, M.; Voipio-Pulkki, L.-M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000, 60, 1789–1792. [Google Scholar] [PubMed]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem. Pharmacol. 2011, 81, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, C.; Kong, C.-Y.; Song, P.; Wu, H.-M.; Xu, S.-C.; Yuan, Y.-P.; Deng, W.; Ma, Z.-G.; Tang, Q.-Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Sun, W.; Zhang, Z.; Zheng, Y. The role of Nrf2-mediated pathway in cardiac remodeling and heart failure. Oxidative Med. Cell. Longev. 2014, 2014, 260429. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Ammar, R.B.; Al-Saeedi, F.J.; Mohamed, M.E.; ElNaggar, M.A.; Al-Ramadan, S.Y.; Bekhet, G.M.; Soliman, A.M. Kaempferol Inhibits Zearalenone-Induced Oxidative Stress and Apoptosis via the PI3K/Akt-Mediated Nrf2 Signaling Pathway: In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2021, 22, 217. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shen, Q.; Liu, G.; Ye, J.; Sun, G.; Sun, X. Myricitrin attenuates high glucose-induced apoptosis through activating Akt-Nrf2 signaling in H9c2 cardiomyocytes. Molecules 2016, 21, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Chen, F.; Lei, J.; Li, Q.; Zhou, B. Activation of the miR-34a-mediated SIRT1/mTOR signaling pathway by urolithin A attenuates D-galactose-induced brain aging in mice. Neurotherapeutics 2019, 16, 1269–1282. [Google Scholar] [CrossRef]
- Seymour, E.M.; Bennink, M.R.; Bolling, S.F. Diet-relevant phytochemical intake affects the cardiac AhR and nrf2 transcriptome and reduces heart failure in hypertensive rats. J. Nutr. Biochem. 2013, 24, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Sun, G.; Wang, J.; Zhang, H.; Sun, X. Naringin protects against anoxia/reoxygenation-induced apoptosis in H9c2 cells via the Nrf2 signaling pathway. Food Funct. 2015, 6, 1331–1344. [Google Scholar] [CrossRef]
- Yu, H.; Chen, B.; Ren, Q. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1α/BNIP3 pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3657–3663. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Zhou, Y.; Xia, Q.; Yao, L.; Chang, X. Astragaloside IV Regulates the PI3K/Akt/HO-1 Signaling Pathway and Inhibits H9c2 Cardiomyocyte Injury Induced by Hypoxia–Reoxygenation. Biol. Pharm. Bull. 2019, 42, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, İ.; Günay, A.E.; Yerer, M.B.; Demirpolat, E.; Doğan, S.; Yay, A.H.; Kafadar, İ.H. Effect of kirenol on the interaction between the WNT/β-Catenin and RUNX2/TCF/LEF1 pathways in fracture healing in vivo. Acta Orthop. Traumatol. Turc. 2020, 54, 320. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wu, J.; Li, Q.; Jin, L.; Qu, Y.; Liang, B.; Zhu, X.; Du, H.; Jie, L. Kirenol inhibit the function and inflammation of fibro-blast-like synoviocytes in rheumatoid arthritis in vitro and in vivo. Front. Immunol. 2019, 10, 1304. [Google Scholar]
- Zhong, Z.; Zhang, Q.; Tao, H.; Sang, W.; Cui, L.; Qiang, W.; San Cheang, W.; Hu, Y.; Yu, H.; Wang, Y. Anti-inflammatory activities of Sigesbeckia glabrescens Makino: Combined in vitro and in silico investigations. Chin. Med. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Yu, Q.-H.; Cheng, Y.; Guo, X.-J. Simultaneous quantification of eight major constituents in Herba Siegesbeckiae by liquid chromatography coupled with electrospray ionization time-of-flight tandem mass spectrometry. J. Pharm. Biomed. Anal. 2011, 55, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Xin, H.-l.; Gao, Z.-f.; Lin, W.; Qian, R. Effect of kirenol on cytokines in serum and apoptosis proteins expression in synoviocytes of adjuvant arthritis rats. Chin. Tradit. Herb. Drugs 2007, 38, 1207. [Google Scholar]
- Kim, M.-B.; Song, Y.; Kim, C.; Hwang, J.-K. Kirenol inhibits adipogenesis through activation of the Wnt/β-catenin signaling pathway in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2014, 445, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiao, J.; Wu, Z.-W.; Wang, Z.-M.; Hu, J.; Fu, H.-Z.; Chen, Y.-Y.; Qian, R.-Q. Kirenol exerts a potent anti-arthritic effect in collagen-induced arthritis by modifying the T cells balance. Phytomedicine 2012, 19, 882–889. [Google Scholar] [CrossRef]
- Manzella, C.; Singhal, M.; Alrefai, W.A.; Saksena, S.; Dudeja, P.K.; Gill, R.K. Serotonin is an endogenous regulator of intestinal CYP1A1 via AhR. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xu, C.; Li, Q.; Gao, X.; Sugano, E.; Tomita, H.; Yang, L.; Shi, S. Thioredoxin 2 offers protection against mitochondrial oxidative stress in H9c2 cells and against myocardial hypertrophy induced by hyperglycemia. Int. J. Mol. Sci. 2017, 18, 1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-L.; Thiyagarajan, V.; Shen, P.-C.; Mathew, D.C.; Lin, K.-Y.; Liao, J.-W.; Hseu, Y.-C. Anti-EMT properties of CoQ0 attributed to PI3K/AKT/NFKB/MMP-9 signaling pathway through ROS-mediated apoptosis. J. Exp. Clin. Cancer Res. 2019, 38, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.-M.; Heublein, D.M.; Perrella, M.A.; Lerman, A.; Rodeheffer, R.J.; McGregor, C.; Edwards, W.D.; Schaff, H.V.; Burnett, J.C., Jr. Natriuretic peptide system in human heart failure. Circulation 1993, 88, 1004–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginauld, S.H.; Cannone, V.; Iyer, S.; Scott, C.; Bailey, K.; Schaefer, J.; Chen, Y.; Sangaralingham, S.J.; Burnett, J.C. Differential regulation of ANP and BNP in acute decompensated heart failure: Deficiency of ANP. JACC Heart Fail. 2019, 7, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Takeshita, S.; Gao, D.; Ishida, T.; Kawashima, S.; Akita, H.; Tawa, R.; Sakurai, H.; Yokoyama, M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis 2001, 155, 45–52. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spallarossa, P.; Altieri, P.; Garibaldi, S.; Ghigliotti, G.; Barisione, C.; Manca, V.; Fabbi, P.; Ballestrero, A.; Brunelli, C.; Barsotti, A. Matrix metalloproteinase-2 and-9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD (P) H oxidase. Cardiovasc. Res. 2006, 69, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Jane-Lise, S.; Corda, S.; Chassagne, C.; Rappaport, L. The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart Fail. Rev. 2000, 5, 239–250. [Google Scholar] [CrossRef]
- Guo, L.; Jiang, X.; Tian, H.-Y.; Yao, S.-J.; Li, B.-Y.; Zhang, R.-J.; Zhang, S.-S.; Sun, X. Detection of BPDE-DNA adducts in human umbilical cord blood by LC-MS/MS analysis. J. Food Drug Anal. 2019, 27, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Childers, R.C.; Sunyecz, I.; West, T.A.; Cismowski, M.J.; Lucchesi, P.A.; Gooch, K.J. Role of the cytoskeleton in the development of a hypofibrotic cardiac fibroblast phenotype in volume overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H596–H608. [Google Scholar] [CrossRef]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Day, C.H.; Lee, N.-H.; Chen, Y.-F.; Yang, J.-J.; Lin, C.-H.; Chen, R.-J.; Rajendran, P.; Viswanadha, V.P.; Huang, C.-Y. Tanshinone IIA inhibits β-catenin nuclear translocation and IGF-2R activation via estrogen receptors to suppress angiotensin II-induced H9c2 cardiomyoblast cell apoptosis. Int. J. Med. Sci. 2017, 14, 1284. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-P.; Sivalingam, K.; Shibu, M.A.; Peramaiyan, R.; Day, C.H.; Shen, C.-Y.; Lai, C.-H.; Chen, R.-J.; Viswanadha, V.P.; Chen, Y.-F. Protective effect of Fisetin against angiotensin II-induced apoptosis by activation of IGF-IR-PI3K-Akt signaling in H9c2 cells and spontaneous hypertension rats. Phytomedicine 2019, 57, 1–8. [Google Scholar] [CrossRef]
- Cao, Y.; Ruan, Y.; Shen, T.; Huang, X.; Li, M.; Yu, W.; Zhu, Y.; Man, Y.; Wang, S.; Li, J. Astragalus polysaccharide suppresses doxorubicin-induced cardiotoxicity by regulating the PI3k/Akt and p38MAPK pathways. Oxidative Med. Cell. Longev. 2014, 2014, 674219. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K. Akt Signaling and Growth of the Heart; American Heart Association: Dallas, TX, USA, 2006. [Google Scholar]
- Hosseini, A.; Bakhtiari, E.; Mousavi, S.H. Protective effect of Hibiscus sabdariffa on doxorubicin-induced cytotoxicity in H9c2 cardiomyoblast cells. Iran. J. Pharm. Res. IJPR 2017, 16, 708. [Google Scholar] [PubMed]
- Zhang, Y.; Ahmad, K.A.; Khan, F.U.; Yan, S.; Ihsan, A.U.; Ding, Q. Chitosan oligosaccharides prevent doxorubicin-induced oxidative stress and cardiac apoptosis through activating p38 and JNK MAPK mediated Nrf2/ARE pathway. Chem. Biol. Interact. 2019, 305, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; Cole, M.P.; St. Clair, D.K.; Ittarat, W.; Szweda, L.I.; Oberley, T.D. Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicol. Pathol. 2004, 32, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Bai, Y.; Jiang, X.; Zhou, S.; Wang, Y.; Wintergerst, K.A.; Cui, T.; Ji, H.; Tan, Y.; Cai, L. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ss/Fyn pathway. Redox Biol. 2018, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Huang, X.Y.; Li, L.; Fan, X.H.; Li, P.C.; Huang, C.Q.; Xiao, J.; Gui, R.; Wang, S. Attenuation of diabetic cardiomyopathy by relying on kirenol to suppress inflammation in a diabetic rat model. J. Cell. Mol. Med. 2019, 23, 7651–7663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Liu, D.; Xing, R.; Song, H.; Tian, X.; Yan, C.; Han, Y. Orosomucoid 1 Attenuates Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes via Nrf2 Signaling. BioMed Res. Int. 2020, 2020, 5923572. [Google Scholar] [CrossRef] [PubMed]
- Sunitha, M.C.; Dhanyakrishnan, R.; PrakashKumar, B.; Nevin, K.G. p-Coumaric acid mediated protection of H9c2 cells from Doxorubicin-induced cardiotoxicity: Involvement of augmented Nrf2 and autophagy. Biomed. Pharmacother. 2018, 102, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-H.; Shibu, M.A.; Peramaiyan, R.; Chen, Y.-F.; Shen, C.-Y.; Hsieh, Y.-L.; Chen, R.-J.; Viswanadha, V.P.; Kuo, W.-W.; Huang, C.-Y. Bioactive flavone fisetin attenuates hypertension associated cardiac hypertrophy in H9c2 cells and in spontaneously hypertension rats. J. Funct. Foods 2019, 52, 212–218. [Google Scholar] [CrossRef]
- Bharathi Priya, L.; Baskaran, R.; Huang, C.Y.; Vijaya Padma, V. Neferine modulates IGF-1R/Nrf2 signaling in doxorubicin treated H9c2 cardiomyoblasts. J. Cell. Biochem. 2018, 119, 1441–1452. [Google Scholar] [CrossRef]
- Ganguly, S.; Chatterjee, E.; Sarkar, S. Targeting Insulin-Like Growth Factor-I receptor signaling pathways improve compromised function during cardiac hypertrophy. J. Integr. Cardiol. 2015. [Google Scholar] [CrossRef]
- Lu, L.; Wu, W.; Yan, J.; Li, X.; Yu, H.; Yu, X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int. J. Cardiol. 2009, 134, 82–90. [Google Scholar] [CrossRef]
- Troncoso, R.; Díaz-Elizondo, J.; Espinoza, S.P.; Navarro-Marquez, M.F.; Oyarzún, A.P.; Riquelme, J.A.; Garcia-Carvajal, I.; Díaz-Araya, G.; García, L.; Hill, J.A. Regulation of cardiac autophagy by insulin-like growth factor 1. IUBMB Life 2013, 65, 593–601. [Google Scholar] [CrossRef]
- Shati, A.A. Doxorubicin-induces NFAT/Fas/FasL cardiac apoptosis in rats through activation of calcineurin and P38 MAPK and inhibition of mTOR signalling pathways. Clin. Exp. Pharmacol. Physiol. 2020, 47, 660–676. [Google Scholar] [CrossRef]
- Spallarossa, P.; Altieri, P.; Barisione, C.; Passalacqua, M.; Aloi, C.; Fugazza, G.; Frassoni, F.; Podestà, M.; Canepa, M.; Ghigliotti, G. p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PLoS ONE 2010, 5, e15583. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Molkentin, J.D. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J. Mol. Cell. Cardiol. 2016, 101, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarmohammadi, F.; Rezaee, R.; Karimi, G. Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/ARE signaling pathway. Phytother. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lyakhovich, A.; Surrallés, J. Constitutive activation of caspase-3 and Poly ADP ribose polymerase cleavage in fanconi anemia cells. Mol. Cancer Res. 2010, 8, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzahrani, A.M.; Rajendran, P.; Veeraraghavan, V.P.; Hanieh, H. Cardiac Protective Effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 Cells through Nrf2 Signaling via PI3K/AKT Pathways. Int. J. Mol. Sci. 2021, 22, 3269. https://doi.org/10.3390/ijms22063269
Alzahrani AM, Rajendran P, Veeraraghavan VP, Hanieh H. Cardiac Protective Effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 Cells through Nrf2 Signaling via PI3K/AKT Pathways. International Journal of Molecular Sciences. 2021; 22(6):3269. https://doi.org/10.3390/ijms22063269
Chicago/Turabian StyleAlzahrani, Abdullah M., Peramaiyan Rajendran, Vishnu Priya Veeraraghavan, and Hamza Hanieh. 2021. "Cardiac Protective Effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 Cells through Nrf2 Signaling via PI3K/AKT Pathways" International Journal of Molecular Sciences 22, no. 6: 3269. https://doi.org/10.3390/ijms22063269
APA StyleAlzahrani, A. M., Rajendran, P., Veeraraghavan, V. P., & Hanieh, H. (2021). Cardiac Protective Effect of Kirenol against Doxorubicin-Induced Cardiac Hypertrophy in H9c2 Cells through Nrf2 Signaling via PI3K/AKT Pathways. International Journal of Molecular Sciences, 22(6), 3269. https://doi.org/10.3390/ijms22063269