
Monomethyl Auristatin E Grafted-Liposomes to Target Prostate Tumor Cell Lines

 , ,
, ,  and
and
Abstract
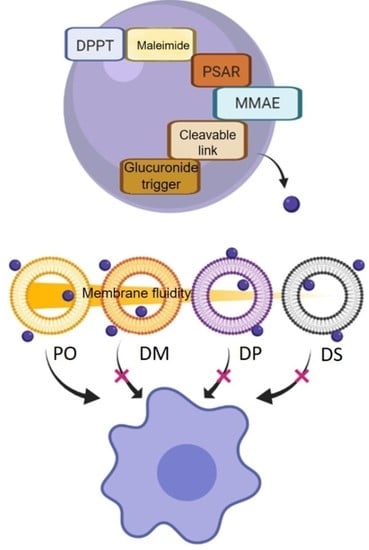
:
1. Introduction
2. Results and Discussion
2.1. DPPT-MMAE Compound Preparation
2.2. Liposome Characterization
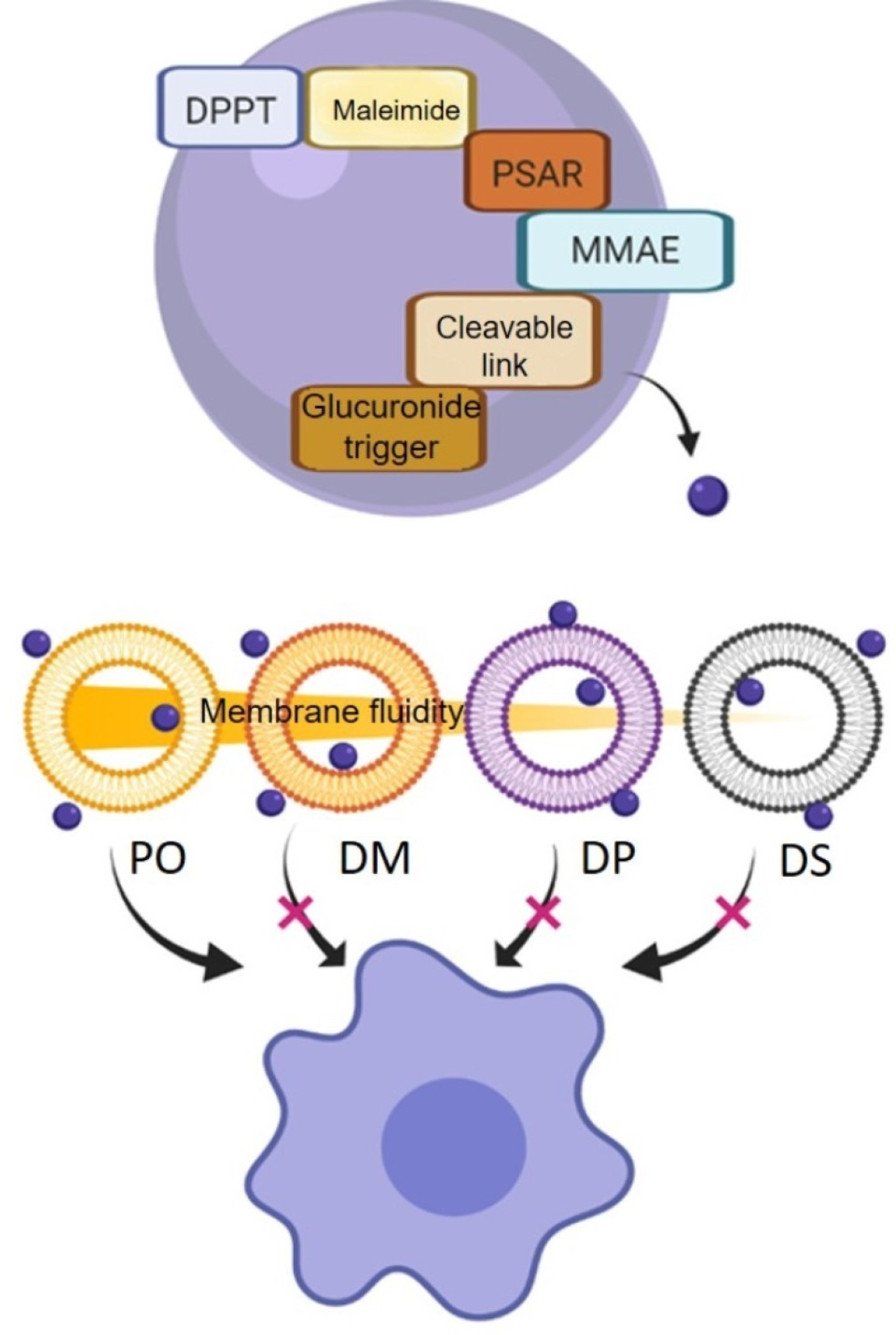
2.3. Liposome Membrane Fluidity
2.4. Liposome Stability over Time
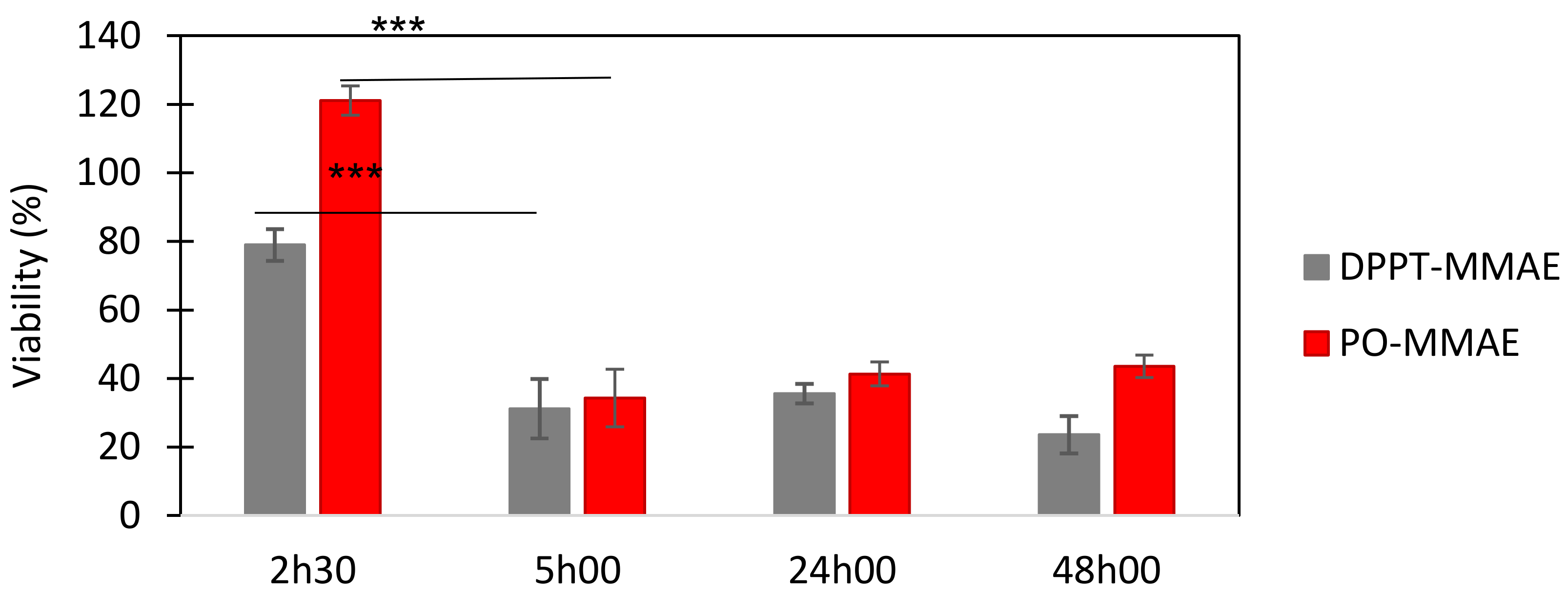
2.5. Liposome-Attached MMAE Effect on PC-3 Prostate Tumor Cells
2.6. Selectivity of Liposomes
3. Materials and Methods
3.1. Synthesis of DPPT-MMAE Derivative
3.2. Liposome Preparation
3.3. Liposome Characterization
3.4. Cell Culture
3.5. Fluorescence Microscopy Experiments
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dacoba, T.G.; Anthiya, S.; Berrecoso, G.; Fernández-Mariño, I.; Fernández-Varela, C.; Crecente-Campo, J.; Teijeiro-Osorio, D.; Torres Andón, F.; Alonso, M.J. Nano-oncologicals: A tortoise trail reaching new avenues. Adv. Funct. Mater. 2021, 2009860, 1–36. [Google Scholar] [CrossRef]
- Pinton, L.; Magri, S.; Masetto, E.; Vettore, M.; Schibuola, I.; Ingangi, V.; Marigo, I.; Matha, K.; Benoit, J.P.; Della Puppa, A.; et al. Targeting of immunosuppressive myeloid cells from glioblastoma patients by modulation of size and surface charge of lipid nanocapsules. J. Nanobiotechnol. 2020, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Matha, K.; Lollo, G.; Taurino, G.; Respaud, R.; Marigo, I.; Shariati, M.; Bussolati, O.; Vermeulen, A.; Remaut, K.; Benoit, J.P. Bioinspired hyaluronic acid and polyarginine nanoparticles for DACHPt delivery. Eur. J. Pharm. Biopharm. 2020, 150, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lollo, G.; Matha, K.; Bocchiardo, M.; Bejaud, J.; Marigo, I.; Virgone-Carlotta, A.; Dehoux, T.; Rivière, C.; Rieu, J.P.; Briançon, S.; et al. Drug delivery to tumours using a novel 5-FU derivative encapsulated into lipid nanocapsules. J. Drug Target. 2019, 27, 634–645. [Google Scholar] [CrossRef]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Webb, M.S.; Harasym, T.O.; Masin, D.; Bally, M.B.; Mayer, L.D. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and therapeutic properties of vincristine in murine and human tumour models. Br. J. Cancer 1995, 72, 896–904. [Google Scholar] [CrossRef]
- Martin, F.; Huang, A.; Uziely, B.; Kaufman, B.; Safra, T. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Forssen, E.A.; Coulter, D.M.; Proffitt, R.T. Selective in Vivo Localization of Daunorubicin Small Unilamellar Vesicles in Solid Tumors. Cancer Res. 1992, 52, 3255–3261. [Google Scholar]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef] [Green Version]
- Balazsovits, J.A.E.; Mayer, L.D.; Bally, M.B.; Cullis, P.R.; McDonell, M.; Ginsberg, R.S.; Falk, R.E. Analysis of the effect of liposome encapsulation on the vesicant properties, acute and cardiac toxicities, and antitumor efficacy of doxorubicin. Cancer Chemother. Pharmacol. 1989, 23, 81–86. [Google Scholar] [CrossRef]
- Chen, J.; He, C.Q.; Lin, A.H.; Gu, W.; Chen, Z.P.; Li, W.; Cai, B.C. Thermosensitive liposomes with higher phase transition temperature for targeted drug delivery to tumor. Int. J. Pharm. 2014, 475, 408–415. [Google Scholar] [CrossRef]
- Hillaireau, H.; Couvreur, P. Nanocarriers’ entry into the cell: Relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66, 2873–2896. [Google Scholar] [CrossRef]
- Ha, K.D.; Bidlingmaier, S.M.; Liu, B. Macropinocytosis exploitation by cancers and cancer therapeutics. Front. Physiol. 2016, 7, 381. [Google Scholar] [CrossRef] [Green Version]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Kobayashi, H.; Turkbey, B.; Watanabe, R.; Choyke, P.L. Cancer drug delivery: Considerations in the rational design of nanosized bioconjugates. Bioconjug. Chem. 2014, 25, 2093–2100. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; What is the appropriate target? Theranostics 2014, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Zhai, G.; Wu, J.; Xiang, G.; Mao, W.; Yu, B.; Li, H.; Piao, L.; Lee, L.J.; Lee, R.J. Preparation, characterization and pharmacokinetics of folate receptor-targeted liposomes for docetaxel delivery. J. Nanosci. Nanotechnol. 2009, 9, 2155–2161. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Lin, Y.; Zhang, X.; Feng, C.; Lu, Y.; Gao, Y.; Dong, C. Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: Enhanced cellular uptake and improved therapeutic effects. Int. J. Nanomed. 2017, 12, 1941–1958. [Google Scholar] [CrossRef] [Green Version]
- Song, X.L.; Ju, R.J.; Xiao, Y.; Wang, X.; Liu, S.; Fu, M.; Liu, J.J.; Gu, L.Y.; Li, X.T.; Cheng, L. Application of multifunctional targeting epirubicin liposomes in the treatment of non-small-cell lung cancer. Int. J. Nanomed. 2017, 12, 7433–7451. [Google Scholar] [CrossRef] [Green Version]
- Raju, A.; Muthu, M.S.; Feng, S.S. Trastuzumab-conjugated vitamin e TPGS liposomes for sustained and targeted delivery of docetaxel. Expert Opin. Drug Deliv. 2013, 10, 747–760. [Google Scholar] [CrossRef]
- Bakowsky, H.; Richter, T.; Kneuer, C.; Hoekstra, D.; Rothe, U.; Bendas, G.; Ehrhardt, C.; Bakowsky, U. Adhesion characteristics and stability assessment of lectin-modified liposomes for site-specific drug delivery. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Hattori, T.; Andoh, T.; Sakai, N.; Yamada, H.; Kameyama, Y.; Ohki, K.; Nozawa, Y. Membrane phospholipid composition and membrane fluidity of human brain tumour: A spin label study. Neurol. Res. 1987, 9, 38–43. [Google Scholar] [CrossRef]
- Sherbet, G.V. Membrane fluidity and cancer metastasis. Exp. Cell Biol. 1989, 57, 198–205. [Google Scholar] [CrossRef]
- Sok, M.; Šentjurc, M.; Schara, M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999, 139, 215–220. [Google Scholar] [CrossRef]
- Kaur, J.; Sanyal, S.N. Alterations in membrane fluidity and dynamics in experimental colon cancer and its chemoprevention by diclofenac. Mol. Cell. Biochem. 2010, 341, 99–108. [Google Scholar] [CrossRef]
- Andoh, Y.; Okazaki, S.; Ueoka, R. Molecular dynamics study of lipid bilayers modeling the plasma membranes of normal murine thymocytes and leukemic GRSL cells. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Zouaoui, J.; Trunfio-Sfarghiu, A.M.; Brizuela, L.; Piednoir, A.; Maniti, O.; Munteanu, B.; Mebarek, S.; Girard-Egrot, A.; Landoulsi, A.; Granjon, T. Multi-scale mechanical characterization of prostate cancer cell lines: Relevant biological markers to evaluate the cell metastatic potential. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 3109–3119. [Google Scholar] [CrossRef]
- Peetla, C.; Vijayaraghavalu, S.; Labhasetwar, V. Biophysics of cell membrane lipids in cancer drug resistance: Implications for drug transport and drug delivery with nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 1686–1698. [Google Scholar] [CrossRef] [Green Version]
- Komizu, Y.; Ueoka, H.; Ueoka, R. Selective accumulation and growth inhibition of hybrid liposomes to human hepatocellular carcinoma cells in relation to fluidity of plasma membranes. Biochem. Biophys. Res. Commun. 2012, 418, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Komizu, Y.; Matsumoto, Y.; Ueoka, R. Membrane targeted chemotherapy with hybrid liposomes for colon tumor cells leading to apoptosis. Bioorganic Med. Chem. Lett. 2006, 16, 6131–6134. [Google Scholar] [CrossRef] [PubMed]
- Bompard, J.; Rosso, A.; Brizuela, L.; Mebarek, S.; Blum, L.J.; Trunfio-Sfarghiu, A.M.; Lollo, G.; Granjon, T.; Girard-Egrot, A.; Maniti, O. Membrane Fluidity as a New Means to Selectively Target Cancer Cells with Fusogenic Lipid Carriers. Langmuir 2020, 36, 5134–5144. [Google Scholar] [CrossRef] [PubMed]
- Cheniour, M.; Gueyrard, D.; Goekjian, P.; Marcillat, O.; Maniti, O.; Vigneron, A.; Ibanez, S.; Granjon, T. Fluorescent Probes and Applications Thereof. European Patent EPO19306175.1, 23 September 2019. [Google Scholar]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and innovation in the manufacturing process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Bai, R.; Petit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.M.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents 337. Synthesis of dolastatin 10 structural modifications. Anticancer Drug Des. 1995, 10, 529–544. [Google Scholar]
- Cunningham, D.; Parajuli, K.R.; Zhang, C.; Wang, G.; Mei, J.; Zhang, Q.; Liu, S.; You, Z. Monomethyl auristatin e phosphate inhibits human prostate cancer growth. Prostate 2016, 76, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-Laford, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Azizi, S.A.; Freedy, A.M.; Cal, P.M.S.D.; Gois, P.M.P.; Bernardes, G.J.L. Construction of homogeneous antibody-drug conjugates using site-selective protein chemistry. Chem. Sci. 2016, 7, 2954–2963. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Kancharla, J.; Kaushik, S.; Ansari, A.; Hossain, S.; Goyal, R.; Pandey, M.; Sivaccumar, J.; Hussain, S.; Sarkar, A.; et al. Development of a facile antibody-drug conjugate platform for increased stability and homogeneity. Chem. Sci. 2017, 8, 2387–2395. [Google Scholar] [CrossRef] [Green Version]
- Bodyak, N.; Yurkovetskiy, A.V. Delivering More Payload (High DAR Adcs); Springer: Berlin/Heidelberg, Germany, 2018; pp. 215–240. [Google Scholar] [CrossRef]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse polysarcosine-based highly-loaded antibody-drug conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef] [Green Version]
- Legigan, T.; Clarhaut, J.; Renoux, B.; Tranoy-Opalinski, I.; Monvoisin, A.; Berjeaud, J.M.; Guilhot, F.; Papot, S. Synthesis and antitumor efficacy of a β-glucuronidase-responsive albumin-binding prodrug of doxorubicin. J. Med. Chem. 2012, 55, 4516–4520. [Google Scholar] [CrossRef]
- Alouane, A.; Labruère, R.; Le Saux, T.; Schmidt, F.; Jullien, L. Self-immolative spacers: Kinetic aspects, structure-property relationships, and applications. Angew. Chem. Int. Ed. 2015, 54, 7492–7509. [Google Scholar] [CrossRef]
- Renoux, B.; Raes, F.; Legigan, T.; Péraudeau, E.; Eddhif, B.; Poinot, P.; Tranoy-Opalinski, I.; Alsarraf, J.; Koniev, O.; Kolodych, S.; et al. Targeting the tumour microenvironment with an enzyme-responsive drug delivery system for the efficient therapy of breast and pancreatic cancers. Chem. Sci. 2017, 8, 3427–3433. [Google Scholar] [CrossRef] [Green Version]
- Christie, R.J.; Fleming, R.; Bezabeh, B.; Woods, R.; Mao, S.; Harper, J.; Joseph, A.; Wang, Q.; Xu, Z.Q.; Wu, H.; et al. Stabilization of cysteine-linked antibody drug conjugates with N-aryl maleimides. J. Control. Release 2015, 220, 660–670. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J.; Tilcock, C.P.S. Lipid polymorphism and the roles of lipids in membranes. Chem. Phys. Lipids 1986, 40, 127–144. [Google Scholar] [CrossRef]
- Israelachvili, J. The science and applications of emulsions—An overview. Colloids Surf. A Physicochem. Eng. Asp. 1994, 91, 1–8. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Aeffner, S.; Reusch, T.; Weinhausen, B.; Salditt, T. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc. Natl. Acad. Sci. USA 2012, 109, E1609–E1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasson, P.M.; Pande, V.S. Control of membrane fusion mechanism by lipid composition: Predictions from ensemble molecular dynamics. PLoS Comput. Biol. 2007, 3, 2228–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.; Grossen, P.; Detampel, P.; Siegfried, S.; Witzigmann, D.; Huwyler, J. Zebrafish as an early stage screening tool to study the systemic circulation of nanoparticulate drug delivery systems in vivo. J. Control. Release 2017, 264, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Gingrich, J.R.; Tucker, J.A.; Walther, P.J.; Day, J.W.; Poulton, S.H.M.; Webb, K.S. Establishment and characterization of a new human prostatic carcinoma cell line (DuPro-1). J. Urol. 1991, 146, 915–919. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar | Liposome Preparation | Acyl Chain Composition | Lipid Name and Structure | Tm |
|---|---|---|---|---|
| Main lipid (79.96 %) | PO | 16:0–18:1 PC | POPC 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine | −4 °C |
| DM | 14:0 PC | DMPC 1,2-dimyristoyl-sn-glycero-3-phosphocholine | 24 °C | |
| DP | 16:0 PC | DPPC 1,2-dipalmitoyl-glycero-3-phosphocholine | 41 °C | |
| DS | 18:0 PC | DSPC 1,2-distearoyl-sn-glycero-3-phosphocholine | 55 °C | |
| Fusogenic lipid (20 %) | All preparations | 18:1 (Δ9-Cis) PE | DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | −16 °C |
| DPPT-MMAE (0.04 %) | All preparations | 16:0 |  | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abawi, A.; Wang, X.; Bompard, J.; Bérot, A.; Andretto, V.; Gudimard, L.; Devillard, C.; Petiot, E.; Joseph, B.; Lollo, G.; et al. Monomethyl Auristatin E Grafted-Liposomes to Target Prostate Tumor Cell Lines. Int. J. Mol. Sci. 2021, 22, 4103. https://doi.org/10.3390/ijms22084103
Abawi A, Wang X, Bompard J, Bérot A, Andretto V, Gudimard L, Devillard C, Petiot E, Joseph B, Lollo G, et al. Monomethyl Auristatin E Grafted-Liposomes to Target Prostate Tumor Cell Lines. International Journal of Molecular Sciences. 2021; 22(8):4103. https://doi.org/10.3390/ijms22084103
Chicago/Turabian StyleAbawi, Ariana, Xiaoyi Wang, Julien Bompard, Anna Bérot, Valentina Andretto, Leslie Gudimard, Chloé Devillard, Emma Petiot, Benoit Joseph, Giovanna Lollo, and et al. 2021. "Monomethyl Auristatin E Grafted-Liposomes to Target Prostate Tumor Cell Lines" International Journal of Molecular Sciences 22, no. 8: 4103. https://doi.org/10.3390/ijms22084103
APA StyleAbawi, A., Wang, X., Bompard, J., Bérot, A., Andretto, V., Gudimard, L., Devillard, C., Petiot, E., Joseph, B., Lollo, G., Granjon, T., Girard-Egrot, A., & Maniti, O. (2021). Monomethyl Auristatin E Grafted-Liposomes to Target Prostate Tumor Cell Lines. International Journal of Molecular Sciences, 22(8), 4103. https://doi.org/10.3390/ijms22084103