
Blood Platelets as an Important but Underrated Circulating Source of TGFβ


Abstract
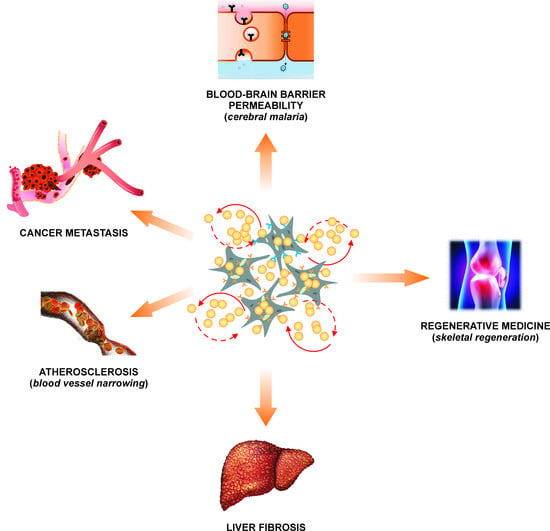
:
{kind=link}
1. Introduction
2. TGFβ in Blood Platelets
3. TGFβ Regulates Platelet Reactivity
4. The Platelet Membrane-Bound TGFβ Pool and Its Possible Involvement in a Modulation of Platelet Reactivity and Platelet-Leukocyte Interactions
5. Measurements of Platelet-Derived TGFβ: Some Methodological Considerations
6. Platelet TGFβ as a Key Component of Platelet-Rich Plasma Used in Regenerative Medicine
7. Platelet TGFβ in Cardiovascular Diseases
8. Platelet TGFβ in Preeclampsia
9. Platelet TGFβ and Liver Diseases
10. Platelet TGFβ in a Cancer
11. Platelet TGFβ in Cerebral Malaria
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-b in human platelets. Identification of major storage site, purification and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [CrossRef]
- Coomes, S.M.; Moore, B.B. Pleiotropic effects of transforming growth factor-β in hematopoietic stem-cell transplantation. Transplantation. 2010, 90, 1139–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family, insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signalling in the kidney, profibrotic and protective effects. Am. J. Physiol. Renal. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012, 119, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Weibrich, G.; Kleis, W.K.; Hafner, G.; Hitzler, W.E. Growth factor levels in platelet-rich plasma and correlations with donor age, sex, and platelet count. J. Craniomaxillofac. Surg. 2002, 30, 97–102. [Google Scholar] [CrossRef]
- Anitua, E.; Sanchez, M.; Nurden, A.T.; Zalduendo, M.; de la Fuente, M.; Azofra, J.; Andia, I. Reciprocal actions of platelet-secreted TGF-beta1 on the production of VEGF and HGF by human tendon cells. Plast. Reconstr. Surg. 2007, 119, 950–959. [Google Scholar] [CrossRef]
- Jin, T.; Almehed, K.; Carlsten, H.; Forsblad-d’Elia, H. Decreased serum levels of TGF-β1 are associated with renal damage in female patients with systemic lupus erythematosus. Lupus 2012, 21, 310–318. [Google Scholar] [CrossRef]
- Lu, R.B.; Lee, S.Y.; Wang, T.Y.; Chang, Y.H.; Chen, P.S.; Yang, Y.K.; Hong, J.S.; Chen, S.L. Long-term heroin use was associated with the downregulation of systemic platelets, BDNF, and TGF-β1, and it contributed to the disruption of executive function in Taiwanese Han Chinese. Drug Alcohol Depend. 2017, 179, 139–145. [Google Scholar] [CrossRef]
- Guo, Y.; Cui, W.; Pei, Y.; Xu, D. Platelets promote invasion and induce epithelial to mesenchymal transition in ovarian cancer cells by TGF-β signalling pathway. Gynecol. Oncol. 2019, 153, 639–650. [Google Scholar] [CrossRef]
- Peraçoli, M.T.; Menegon, F.T.; Borges, V.T.; de Araújo Costa, R.A.; Thomazini-Santos, I.A.; Peraçoli, J.C. Platelet aggregation and TGF-beta(1) plasma levels in pregnant women with preeclampsia. J. Reprod. Immunol. 2008, 79, 79–84. [Google Scholar] [CrossRef]
- Pircher, R.; Jullien, P.; Lawrence, D.A. Beta-transforming growth factor is stored in human blood platelets as a latent high molecular weight complex. Biochem. Biophys. Res. Commun. 1986, 136, 30–37. [Google Scholar] [CrossRef]
- Okada, F.; Yamaguchi, K.; Ichihara, A.; Nakamura, T. Purification and structural analysis of a latent form of transforming growth factor-beta from rat platelets. J. Biochem. 1989, 106, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Hellman, U.; Ten Dijke, P.; Grimsby, S.; Ichijo, H.; Morén, A.; Miyazono, K.; Heldin, C.H. Latent transforming growth factor-beta complex in Chinese hamster ovary cells contains the multifunctional cysteine-rich fibroblast growth factor receptor, also termed E-selectin-ligand or MG-160. Biochem. J. 1997, 324, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Grainger, D.J.; Wakefield, L.; Bethell, J.W.H.; Farndale, W.R.; Metcalfe, C.J. Release and activation of platelet TGF-beta in blood clots during dissolution with plasmin. Nat. Med. 1995, 1, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Blakytny, R.; Ludlow, A.; Martin, G.E.; Ireland, G.; Lund, L.R.; Ferguson, M.W.; Brunner, G. Latent TGF-beta1 activation by platelets. J. Cell Physiol. 2004, 199, 67–76. [Google Scholar] [CrossRef]
- Harrison, S.; Vavken, P.; Kevy, S.; Jacobson, M.; Zurakowski, D.; Murray, M.M. Platelet activation by collagen provides sustained release of anabolic cytokines. Am. J. Sports Med. 2011, 39, 729–734. [Google Scholar] [CrossRef]
- Rath, D.; Chatterjee, M.; Holtkamp, A.; Tekath, N.; Borst, O.; Vogel, S.; Müller, K.; Gawaz, M.; Geisler, T. Evidence of an interaction between TGF-β1 and the SDF-1/CXCR4/CXCR7 axis in human platelets. Thromb. Res. 2016, 144, 79–84. [Google Scholar] [CrossRef]
- Garnica, M.R.; Souto, J.T.; Silva, J.S.; de Andrade, H.F., Jr. Stromal cell derived factor 1 synthesis by spleen cells in rodent malaria, and the effects of in vivo supplementation of SDF-1alpha and CXCR4 receptor blocker. Immunol. Lett. 2002, 83, 47–53. [Google Scholar] [CrossRef]
- Schanz, A.; Winn, V.D.; Fisher, S.J.; Blumenstein, M.; Heiss, C.; Hess, A.P.; Kruessel, J.S.; Mcmaster, M.; North, R.A. Pre-eclampsia is associated with elevated CXCL12 levels in placental syncytiotrophoblasts and maternal blood. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Hoh, B.L.; Hosaka, K.; Downes, D.P.; Nowicki, K.W.; Wilmer, E.N.; Velat, G.J.; Scott, E.W. Stromal cell-derived factor-1 promoted angiogenesis and inflammatory cell infiltration in aneurysm walls. J. Neurosurg. 2014, 120, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Duda, D.G. Role of stromal cell-derived factor 1α pathway in bone metastatic prostate cancer. J. Biomed. Res. 2016, 30, 181–185. [Google Scholar] [PubMed] [Green Version]
- Jackson, E.K.; Zhang, Y.; Gillespie, D.D.; Zhu, X.; Cheng, D.; Jackson, T.C. SDF-1α (Stromal Cell-Derived Factor 1α) Induces Cardiac Fibroblasts, Renal Microvascular Smooth Muscle Cells, and Glomerular Mesangial Cells to Proliferate, Cause Hypertrophy, and Produce Collagen. J. Am. Heart Assoc. 2017, 6, e007253. [Google Scholar] [CrossRef] [Green Version]
- Lev, P.R.; Salim, J.P.; Marta, R.F.; Osorio, M.J.; Goette, N.P.; Molinas, F.C. Platelets possess functional TGF-beta receptors and Smad2 protein. Platelets 2007, 18, 35–42. [Google Scholar] [CrossRef]
- Itoh, S.; Itoh, F.; Goumans, M.J.; Dijke, P. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 2000, 267, 6954–6967. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P. Białka z rodziny SMAD, współczesna wiedza na temat ich ekspresji i potencjalnej roli w chorobachnowotworowych [SMAD family proteins, the current knowledge on their expression and potential role in neoplastic diseases]. Postepy Hig. Med. Dosw. 2014, 68, 301–309. [Google Scholar] [CrossRef]
- Albers, R.E.; Selesniemi, K.; Natale, D.R.C.; Brown, T.L. TGF-β induces Smad2 Phosphorylation, ARE Induction, and Trophoblast Differentiation. Int. J. Stem Cells 2018, 11, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Lannan, K.L.; Sahler, J.; Kim, N.; Spinelli, S.L.; Maggirwar, S.B.; Garraud, O.; Cognasse, F.; Blumberg, N.; Phipps, R.P. Breaking the mold, transcription factors in the anucleate platelet and platelet-derived microparticles. Front. Immunol. 2015, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Solanilla, A.; Villeneuve, J.; Auguste, P.; Hugues, M.; Alioum, A.; Lepreux, S.; Ducroix, J.P.; Duhaut, P.; Conri, C.; Viallard, J.F.; et al. The transport of high amounts of vascular endothelial growth factor by blood platelets underlines their potential contribution in systemic sclerosis angiogenesis. Rheumatology 2009, 48, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [Green Version]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell. Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.; Gaudet, M.; Plesa, M.; Allakhverdi, Z.; Mogas, A.K.; Audusseau, S.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Ludwig, M.S.; et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir. Res. 2019, 20, 234. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, W.; Huang, X.; Cao, A.T.; Bilotta, A.J.; Xiao, Y.; Sun, M.; Chen, L.; Ma, C.; Liu, X.; et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J. Immunol. 2018, 201, 2492–2501. [Google Scholar] [CrossRef]
- Kral, J.B.; Schrottmaier, W.C.; Salzmann, M.; Assinger, A. Platelet Interaction with Innate Immune Cells. Transfus. Med. Hemother. 2016, 43, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passacquale, G.; Vamadevan, P.; Pereira, L.; Hamid, C.; Corrigall, V.; Ferro, A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS ONE 2011, 6, e25595. [Google Scholar] [CrossRef] [Green Version]
- Inui, M.; Tazawa, K.; Kishi, Y.; Takai, T. Platelets convert peripheral blood circulating monocytes to regulatory cells via immunoglobulin G and activating-type Fcγ receptors. BMC Immunol. 2015, 16, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haribhai, D.; Luo, X.; Chen, J.; Jia, S.; Shi, L.; Schroeder, J.A.; Weiler, H.; Aster, R.H.; Hessner, M.J.; Hu, J.; et al. TGF-β1 along with other platelet contents augments Treg cells to suppress anti-FVIII immune responses in hemophilia A mice. Blood Adv. 2016, 1, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.W.; Du, Y.; Liu, X. Platelet-derived TGF-β1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum. Reprod. 2016, 31, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.G.; Read, M.S.; Shermer, R.W. Comparison of certain functions of human platelets separated from blood by various means. Am. J. Pathol. 1974, 76, 323–332. [Google Scholar]
- Walkowiak, B.; Kralisz, U.; Michalec, L.; Majewska, E.; Koziolkiewicz, W.; Ligocka, A.; Cierniewski, C.S. Comparison of platelet aggregability and P-selectin surface expression on platelets isolated by different methods. Thromb. Res. 2000, 99, 495–502. [Google Scholar] [CrossRef]
- Kropf, J.; Schurek, J.O.; Wollner, A.; Gressner, A.M. Immunological measurement of transforming growth factor-beta 1 (TGF-beta1) in blood, assay development and comparison. Clin. Chem. 1997, 43, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Grande, J.P. Role of transforming growth factor-beta in tissue injury and repair. Proc. Soc. Exp. Biol. Med. 1997, 214, 27–40. [Google Scholar] [CrossRef]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-β family in wound healing, burns and scarring, a review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar]
- Dugrillon, A.; Eichler, H.; Kern, S.; Klüter, H. Autologous concentrated platelet-rich plasma (cPRP) for local application in bone regeneration. Int. J. Oral. Maxillofac. Surg. 2002, 31, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Sutter, W.W.; Kaneps, A.J.; Bertone, A.L. Comparison of hematologic values and transforming growth factor-beta and insulin-like growth factor concentrations in platelet concentrates obtained by use of buffy coat and apheresis methods from equine blood. Am. J. Vet. Res. 2004, 65, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Weibrich, G.; Kleis, W.K.; Hitzler, W.E.; Hafner, G. Comparison of the platelet concentrate collection system with the plasma-rich-in-growth-factors kit to produce platelet-rich plasma, a technical report. Int. J. Oral. Maxillofac. Implants 2005, 20, 118–123. [Google Scholar] [PubMed]
- Yang, H.; Yuan, C.; Wu, C.; Qian, J.; Shi, Q.; Li, X.; Zhu, X.; Zou, J. The role of TGF-β1/Smad2/3 pathway in platelet-rich plasma in retarding intervertebral disc degeneration. J. Cell Mol. Med. 2016, 20, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Giovanini, A.F.; Gonzaga, C.C.; Zielak, J.C.; Deliberador, T.M.; Kuczera, J.; Göringher, I.; de Oliveira Filho, M.A.; Baratto-Filho, F.; Urban, C.A. Platelet-rich plasma (PRP) impairs the craniofacial bone repair associated with its elevated TGF-β levels and modulates the co-expression between collagen III and α-smooth muscle actin. J. Orthop. Res. 2011, 29, 457–463. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signalling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, K.T.I.; Seidel, H.; Pujol, C.; Meierhofer, C.; Röschenthaler, F.; Pressler, A.; Stöckl, A.; Nagdyman, N.; Neidenbach, R.C.; von Hundelshausen, P.; et al. Hemostatic abnormalities in adult patients with Marfan syndrome. Cardiovasc. Diagn. Ther. 2019, 9, S209–S220. [Google Scholar] [CrossRef]
- Wang, W.; Vootukuri, S.; Meyer, A.; Ahamed, J.; Coller, B.S. Association between shear stress and platelet-derived transforming growth factor-β1 release and activation in animal models of aortic valve stenosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1924–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Johnston, T.P.; Johansson, D.; Parini, P.; Funa, K.; Svensson, J.; Hansson, G.K. Hypercholesterolemia leads to elevated TGF-beta1 activity and T helper 3-dependent autoimmune responses in atherosclerotic mice. Atherosclerosis 2009, 204, 381–387. [Google Scholar] [CrossRef]
- Aihara, K.; Ikeda, Y.; Yagi, S.; Akaike, M.; Matsumoto, T. Transforming Growth Factor-β1 as a Common Target Molecule for Development of Cardiovascular Diseases, Renal Insufficiency and Metabolic Syndrome. Cardiol. Res. Pract. 2010, 2011, 175381. [Google Scholar] [CrossRef] [Green Version]
- Boccardo, P.; Remuzzi, G.; Galbusera, M. Platelet dysfunction in renal failure. Semin. Thromb. Hemost. 2004, 30, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Bush, R.L.; Yao, Q.; Lumsden, A.B.; Chen, C. Evaluation of platelet deposition and neointimal hyperplasia of heparin-coated small-caliber ePTFE grafts in a canine femoral artery bypass model. J. Surg. Res. 2004, 118, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Icli, A.; Aksoy, F.; Dogan, A.; Arslan, A.; Akcay, S.; Yücel, H.; Ersoy, I.; Gorgulu, O. Increased mean platelet volume in hypertrophic cardiomyopathy. Angiology 2014, 65, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Suslova, T.E.; Sitozhevskii, A.V.; Ogurkova, O.N.; Kravchenko, E.S.; Kologrivova, I.V.; Anfinogenova, Y.; Karpov, R.S. Platelet hemostasis in patients with metabolic syndrome and type 2 diabetes mellitus, cGMP- and NO-dependent mechanisms in the insulin-mediated platelet aggregation. Front. Physiol. 2015, 5, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naicker, T.; Khedun, S.M.; Moodley, J. Transforming growth factor beta(1) levels in platelet depleted plasma in African women with pre-eclampsia. J. Obstet. Gynaecol. 2002, 22, 279–282. [Google Scholar] [CrossRef]
- Muy-Rivera, M.; Sanchez, S.E.; Vadachkoria, S.; Qiu, C.; Bazul, V.; Williams, M.A. Transforming growth factor-beta1 (TGF-beta1) in plasma is associated with preeclampsia risk in Peruvian women with systemic inflammation. Am. J. Hypertens 2004, 17, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Enquobahrie, D.A.; Williams, M.A.; Qiu, C.; Woelk, G.B.; Mahomed, K. Maternal plasma transforming growth factor-beta1 concentrations in preeclamptic and normotensive pregnant Zimbabwean women. J. Matern. Fetal. Neonatal. Med. 2005, 17, 343–348. [Google Scholar] [CrossRef]
- Adu-Gyamfi, E.A.; Lamptey, J.; Duan, F.; Wang, Y.X.; Ding, Y.B. The transforming growth factor β superfamily as possible biomarkers of preeclampsia, a comprehensive review. Biomark. Med. 2019, 13, 1321–1330. [Google Scholar] [CrossRef]
- Clausen, T.; Djurovic, S.; Reseland, J.E.; Berg, K.; Drevon, C.A.; Henriksen, T. Altered plasma concentrations of leptin, transforming growth factor-beta(1) and plasminogen activator inhibitor type 2 at 18 weeks of gestation in women destined to develop pre-eclampsia. Circulating markers of disturbed placentation? Placenta 2002, 23, 380–385. [Google Scholar] [CrossRef]
- Ito, Y.; Abril, E.R.; Bethea, N.W.; McCuskey, M.K.; Cover, C.; Jaeschke, H.; McCuskey, R.S. Mechanisms and pathophysiological implications of sinusoidal endothelial cell gap formation following treatment with galactosamine/endotoxin in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G211–G218. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Hynes, R.O. Extracellular matrix proteins in hemostasis and thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [PubMed] [Green Version]
- Ahamed, J.; Laurence, J. Role of Platelet-Derived Transforming Growth Factor-β1 and Reactive Oxygen Species in Radiation-Induced Organ Fibrosis. Antioxid. Redox Signal. 2017, 27, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Braet, F.; Shleper, M.; Paizi, M.; Brodsky, S.; Kopeiko, N.; Resnick, N.; Spira, G. Liver sinusoidal endothelial cell modulation upon resection and shear stress in vitro. Comp. Hepatol. 2004, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, J.; Burg, N.; Yoshinaga, K.; Janczak, C.A.; Rifkin, D.B.; Coller, B.S. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood 2008, 112, 3650–3660. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, S.M.; Poczatek, M.; Schultz-Cherry, S.; Villain, M.; Murphy-Ullrich, J.E. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J. Biol. Chem. 1999, 274, 13586–13593. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, J.; Janczak, C.A.; Wittkowski, K.M.; Coller, B.S. In vitro and in vivo evidence that thrombospondin-1 (TSP-1) contributes to stirring- and shear-dependent activation of platelet-derived TGF-beta1. PLoS ONE 2009, 4, e6608. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Kirita, A.; Kondo, W.; Matsuura, T.; Nagatsuma, K.; Dohmae, N.; Ogawa, S.; Imajoh-Ohmi, S.; Friedman, S.L.; Rifkin, D.B.; et al. LAP degradation product reflects plasma kallikrein-dependent TGF-β activation in patients with hepatic fibrosis. Springerplus 2014, 3, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells, Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, T.; Ohkohchi, N. Platelets in liver disease, cancer and regeneration. World J. Gastroenterol. 2017, 23, 3228–3239. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E.; Schultz-Cherry, S.; Höök, M. Transforming growth factor-beta complexes with thrombospondin. Mol. Biol. Cell 1992, 3, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Poczatek, M. Activation of latent TGF-beta by thrombospondin-1, mechanisms and physiology. Cytokine Growth Factor Rev. 2000, 11, 59–69. [Google Scholar] [CrossRef]
- Adler, H.L.; McCurdy, M.; Kattan, M.W.; Timme, T.L.; Scardino, P.T.; Thompson, T.C. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J. Urol. 1999, 161, 182–187. [Google Scholar] [CrossRef]
- Baselga, J.; Rothenberg, M.L.; Seoane, J.T.; Daly, T.; Cleverly, A.; Berry, B.; Rhoades, S.K.; Ray, C.A.; Fill, J.; Farrington, D.L.; et al. TGF-beta signalling-related markers in cancer patients with bone metastasis. Biomarkers 2008, 13, 217–236. [Google Scholar] [CrossRef]
- Shariat, S.F.; Walz, J.; Roehrborn, C.G.; Zlotta, A.R.; Perrotte, P.; Suardi, N.; Saad, F.; Karakiewicz, P.I. External validation of a biomarker-based preoperative nomogram predicts biochemical recurrence after radical prostatectomy. J. Clin. Oncol. 2008, 26, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Kawasumi, M.; Kitoh, H.; Siwicka, K.A.; Ishiguro, N. The Effect of the Platelet Concentration in Platelet-Rich Plasma Gel on the Regeneration of Bone. J. Bone Jt. Surg. Br. 2007, 90-B, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Uggeri, J.; Belletti, S.; Guizzardi, S.; Poli, T.; Cantarelli, S.; Scandroglio, R.; Gatti, R. Dose-Dependent Effects of Platelet Gel Releasate on Activites of Human Osteoblasts. J. Periodontol. 2007, 78, 1985–1991. [Google Scholar] [CrossRef]
- Nair, M.; Varma, H.K.; John, A. Platelet-Rich Plasma and Fibrin Glue-Coated Bioactive Ceramics Enhance Growth and Differentiation of Goat Bone Marrow-Derived Stem Cells. Tissue Eng. Part A 2009, 15, 1619–1631. [Google Scholar] [CrossRef]
- Karreth, F.G.; Fischer, M.B.; Watzek, G. Platelet-Released Supernatants Stimulate Formation of Osteoclast-like Cells through a Prostaglandin/RANKL-Dependent Mechanism. Bone 2002, 30, 726–732. [Google Scholar]
- Kerr, B.A.; McCabe, N.P.; Feng, W.; Byzova, T.V. Platelets govern pre-metastatic tumour communication to bone. Oncogene 2013, 32, 4319–4324. [Google Scholar] [CrossRef] [Green Version]
- Kerr, B.A.; Miocinovic, R.; Smith, A.K.; Klein, E.A.; Byzova, T.V. Comparison of tumour and microenvironment secretomes in plasma and in platelets during prostate cancer growth in a xenograft model. Neoplasia 2010, 12, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Kerr, B.A.; Harris, K.S.; Shi, L.; Willey, J.S.; Soto-Pantoja, D.R.; Byzova, T.V. Platelet TSP-1 Controls Prostate Cancer-Induced Osteoclast Differentiation and Bone Marrow-Derived Cell Mobilization through TGFβ-1. Am. J. Clin. Exp. Urol. 2021, 9, 18. [Google Scholar]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signalling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inyang, A.L.; Sodeinde, O.; Okpako, D.T.; Essien, E.M. Platelet reactions after interaction with cultured Plasmodium falciparum infected erythrocytes. Br. J. Haematol. 1987, 66, 375–378. [Google Scholar] [CrossRef]
- Srivastava, K.; Cockburn, I.A.; Swaim, A.; Thompson, L.E.; Tripathi, A.; Fletcher, C.A.; Shirk, E.M.; Sun, H.; Kowalska, M.A.; Fox-Talbot, K.; et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell. Host Microbe 2008, 4, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Asare, R.; Opoku-Okrah, C.; Danquah, K.O.; Opare-Sem, O.; Addai-Mensah, O.; Gyamfi, D.; Amponsah, F.A.; Afriyie, E.Y.; Duneeh, R.V.; Ofosu, D.N.; et al. Assessment of platelet indices and platelet activation markers in children with Plasmodium falciparum malaria. Malar. J. 2020, 19, 143. [Google Scholar] [CrossRef] [Green Version]
- Tanahashi, N.; Fukuuchi, Y.; Tomita, M.; Tomita, Y.; Inoue, K.; Satoh, H.; Abe, T. Adhesion of adenosine diphosphate-activated platelets to human brain microvascular endothelial cells under flow in vitro is mediated via GPIIb/IIIa. Neurosci. Lett. 2001, 301, 33–36. [Google Scholar] [CrossRef]
- Pignatelli, P.; De Biase, L.; Lenti, L.; Tocci, G.; Brunelli, A.; Cangemi, R.; Riondino, S.; Grego, S.; Volpe, M.; Violi, F. Tumor necrosis factor-alpha as trigger of platelet activation in patients with heart failure. Blood 2005, 106, 1992–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, J.; Merkle, M.; Wörnle, M.; Ribeiro, A.; Czermak, T.; Stampnik, Y.; Mannell, H.; Niemeyer, M.; Vielhauer, V.; Krötz, F. Prothrombotic effects of tumour necrosis factor alpha in vivo are amplified by the absence of TNF-alpha receptor subtype 1 and require TNF-alpha receptor subtype 2. Arthritis Res. Ther. 2012, 14, R225. [Google Scholar] [CrossRef] [Green Version]
- Wassmer, S.C.; de Souza, J.B.; Frère, C.; Candal, F.J.; Juhan-Vague, I.; Grau, G.E. TGF-beta1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis, a new mechanism for microvascular lesion during cerebral malaria. J. Immunol. 2006, 176, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Ramirez, M.A.; Fischer, R.; Torres-Badillo, C.C.; Davies, H.A.; Logan, K.; Pfizenmaier, K.; Male, D.K.; Sharrack, B.; Romero, I.A. Role of caspases in cytokine-induced barrier breakdown in human brain endothelial cells. J. Immunol. 2012, 189, 3130–3139. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Kondo, T.; Chan, P.H. Blood-brain barrier disruption, edema formation, and apoptotic neuronal death following cold injury. Acta Neurochir. Suppl. 1997, 70, 234–236. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. Int. J. Mol. Sci. 2021, 22, 4492. https://doi.org/10.3390/ijms22094492
Karolczak K, Watala C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. International Journal of Molecular Sciences. 2021; 22(9):4492. https://doi.org/10.3390/ijms22094492
Chicago/Turabian StyleKarolczak, Kamil, and Cezary Watala. 2021. "Blood Platelets as an Important but Underrated Circulating Source of TGFβ" International Journal of Molecular Sciences 22, no. 9: 4492. https://doi.org/10.3390/ijms22094492
APA StyleKarolczak, K., & Watala, C. (2021). Blood Platelets as an Important but Underrated Circulating Source of TGFβ. International Journal of Molecular Sciences, 22(9), 4492. https://doi.org/10.3390/ijms22094492