
Effect of Posttranslational Modifications on the Structure and Activity of FTO Demethylase

 ,
,  , , , , , , and
, , , , , , and
Abstract
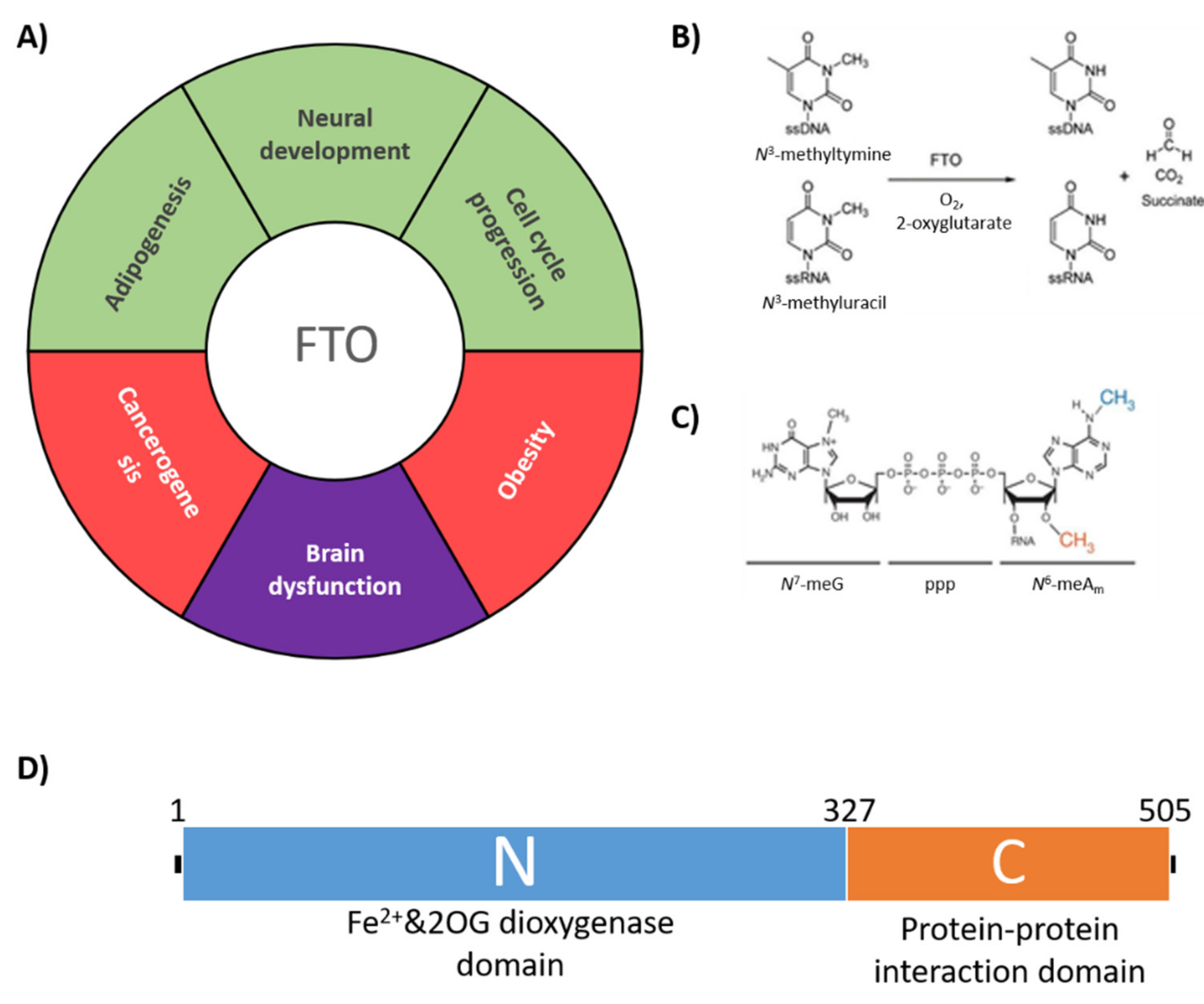
:1. Introduction
2. Results
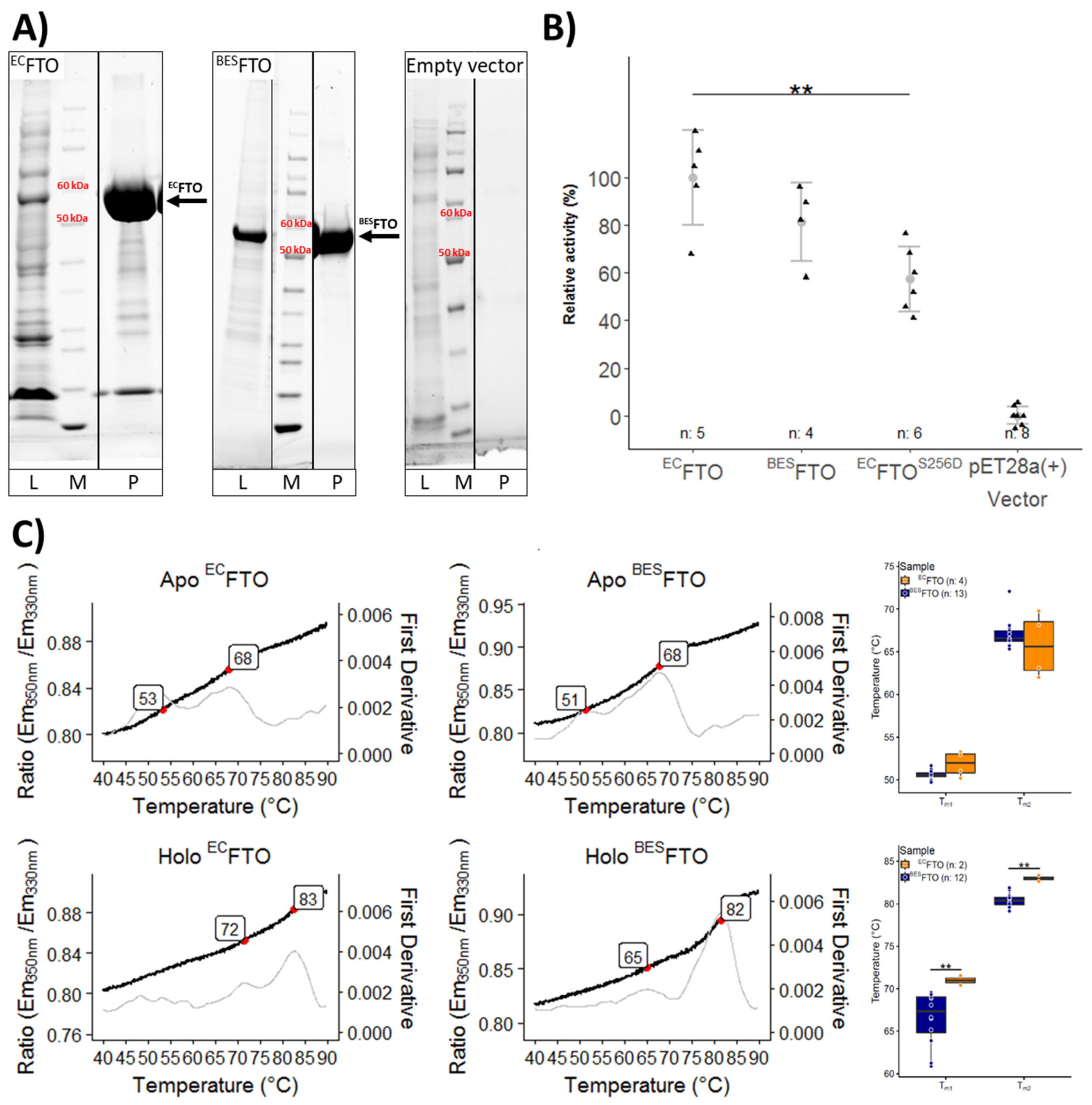
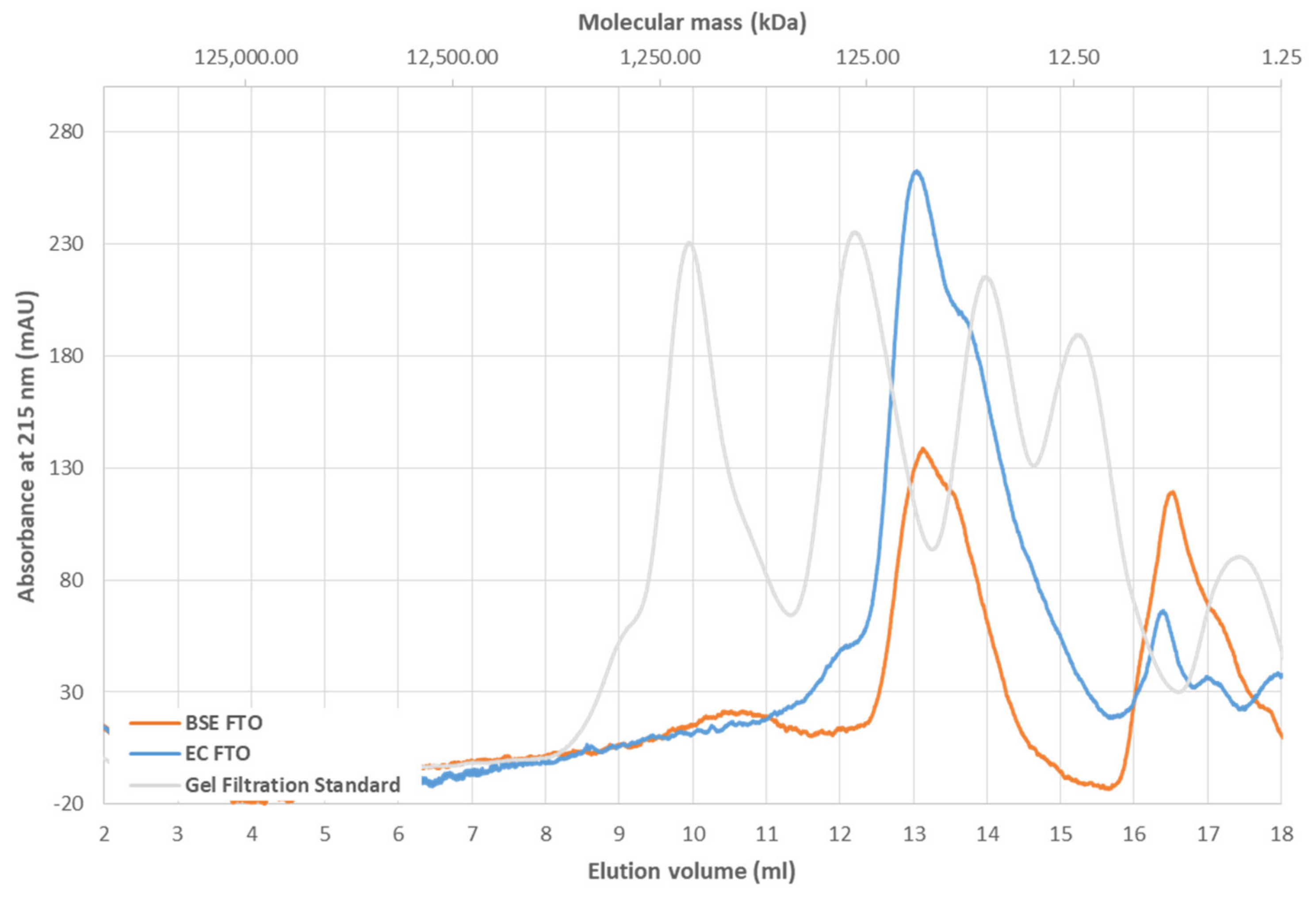
2.1. Thermal Stability of FTO Depends on Expression System Used
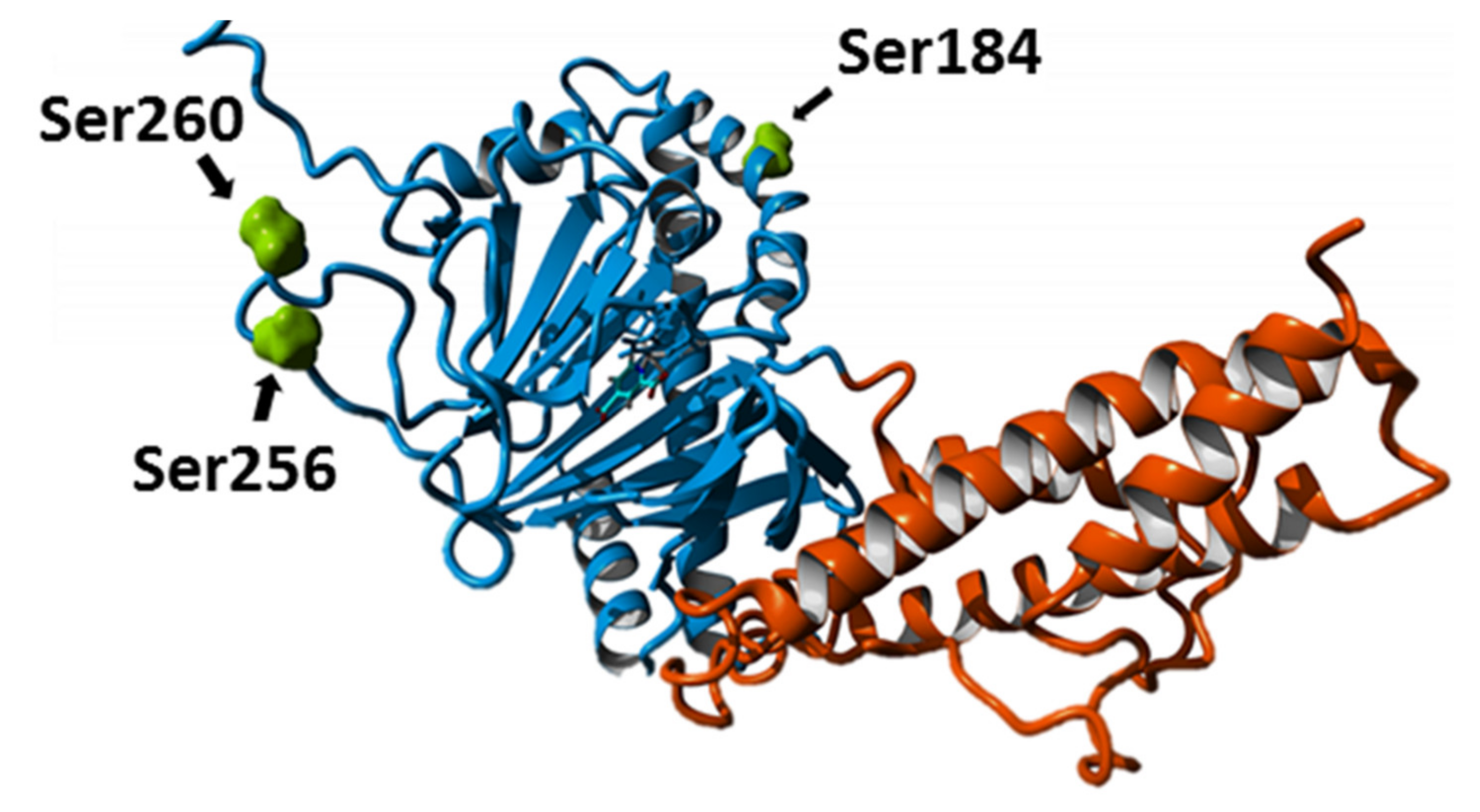
2.2. Posttranslational Modifications Affect FTO Demethylating Activity
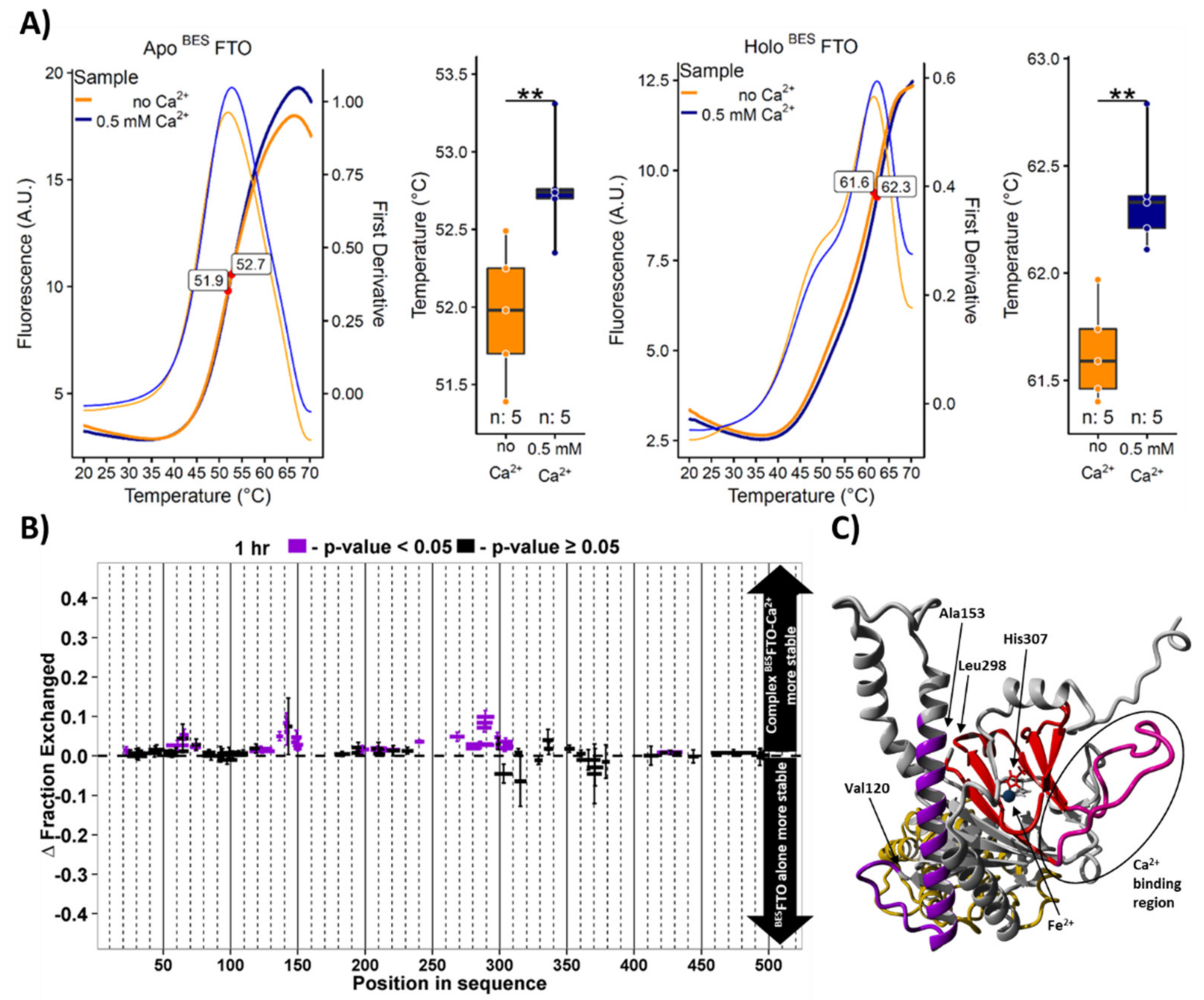
2.3. Calcium Affects FTO Stability
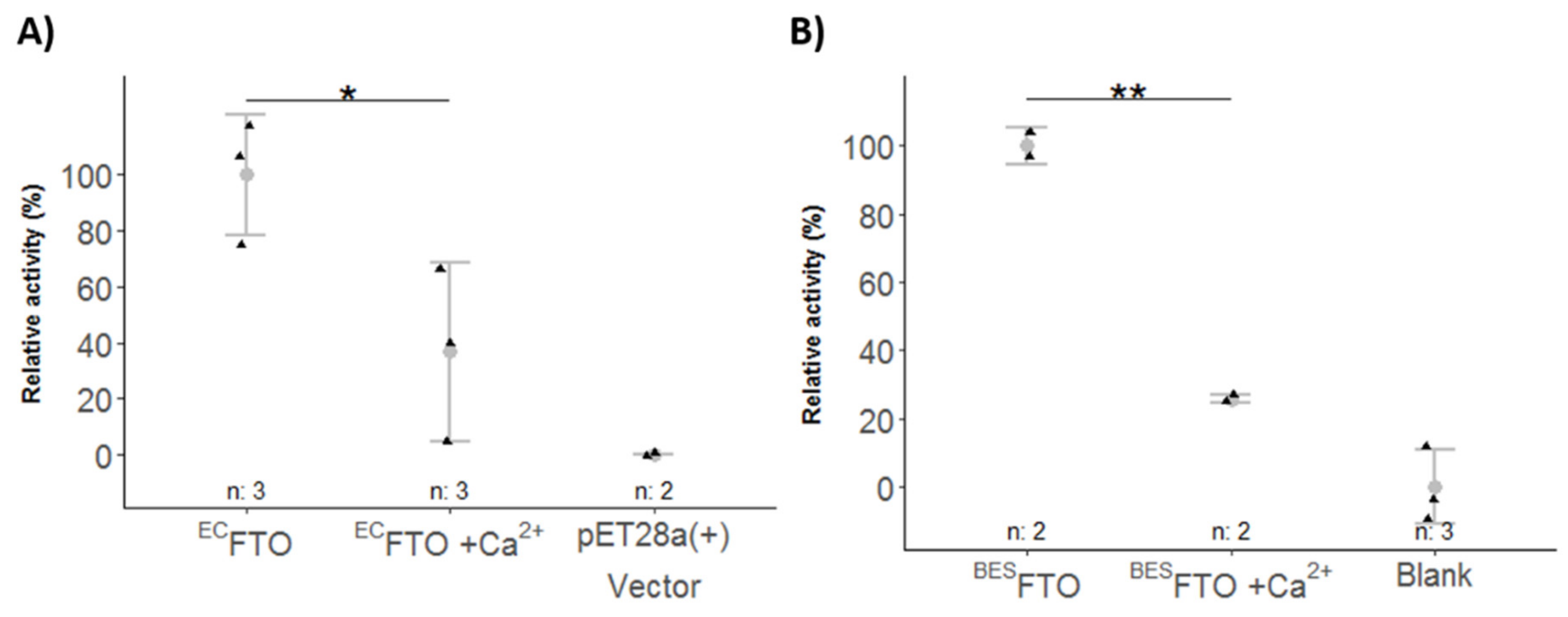
2.4. Ca2+ Affects FTO Catalytic Activity
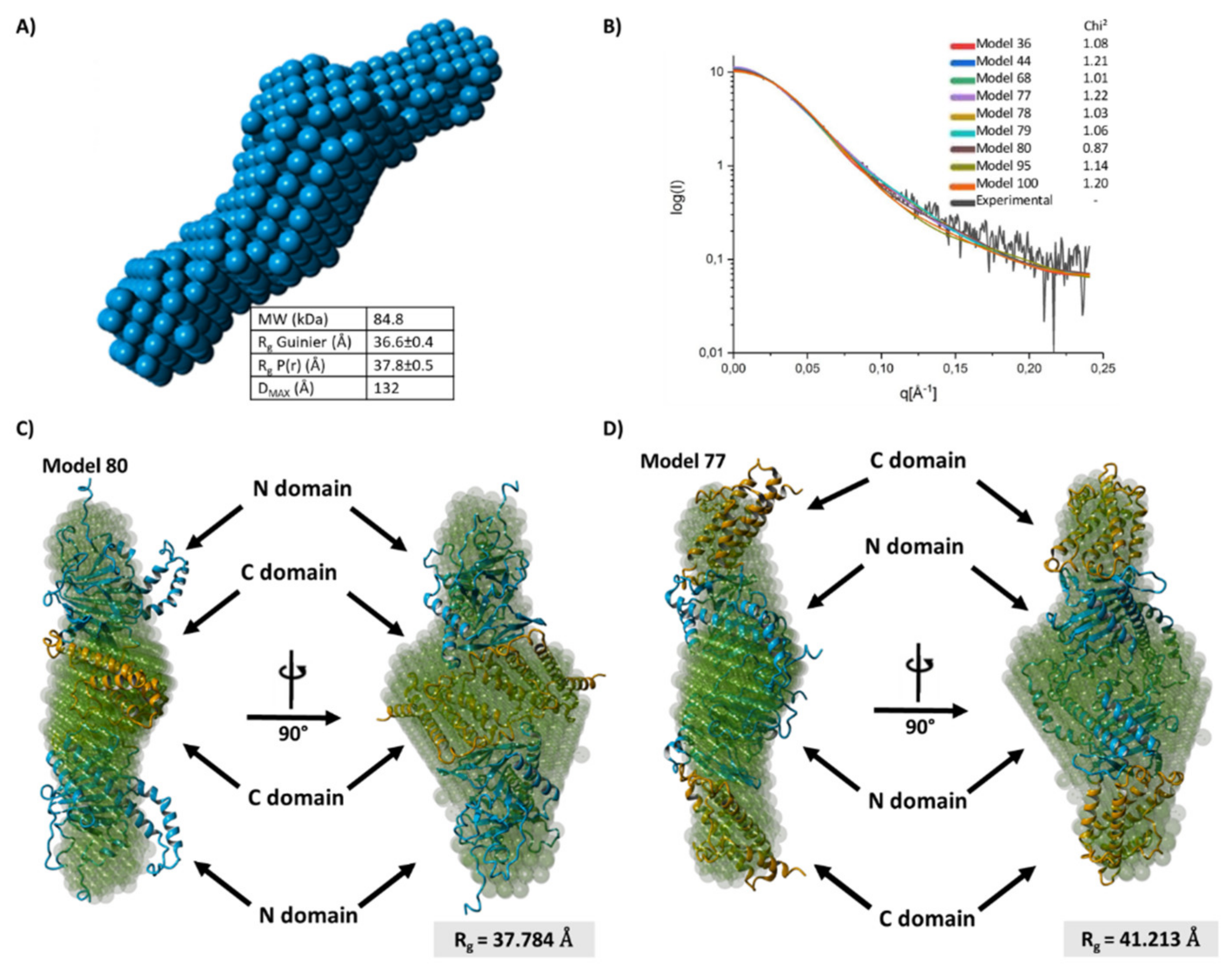
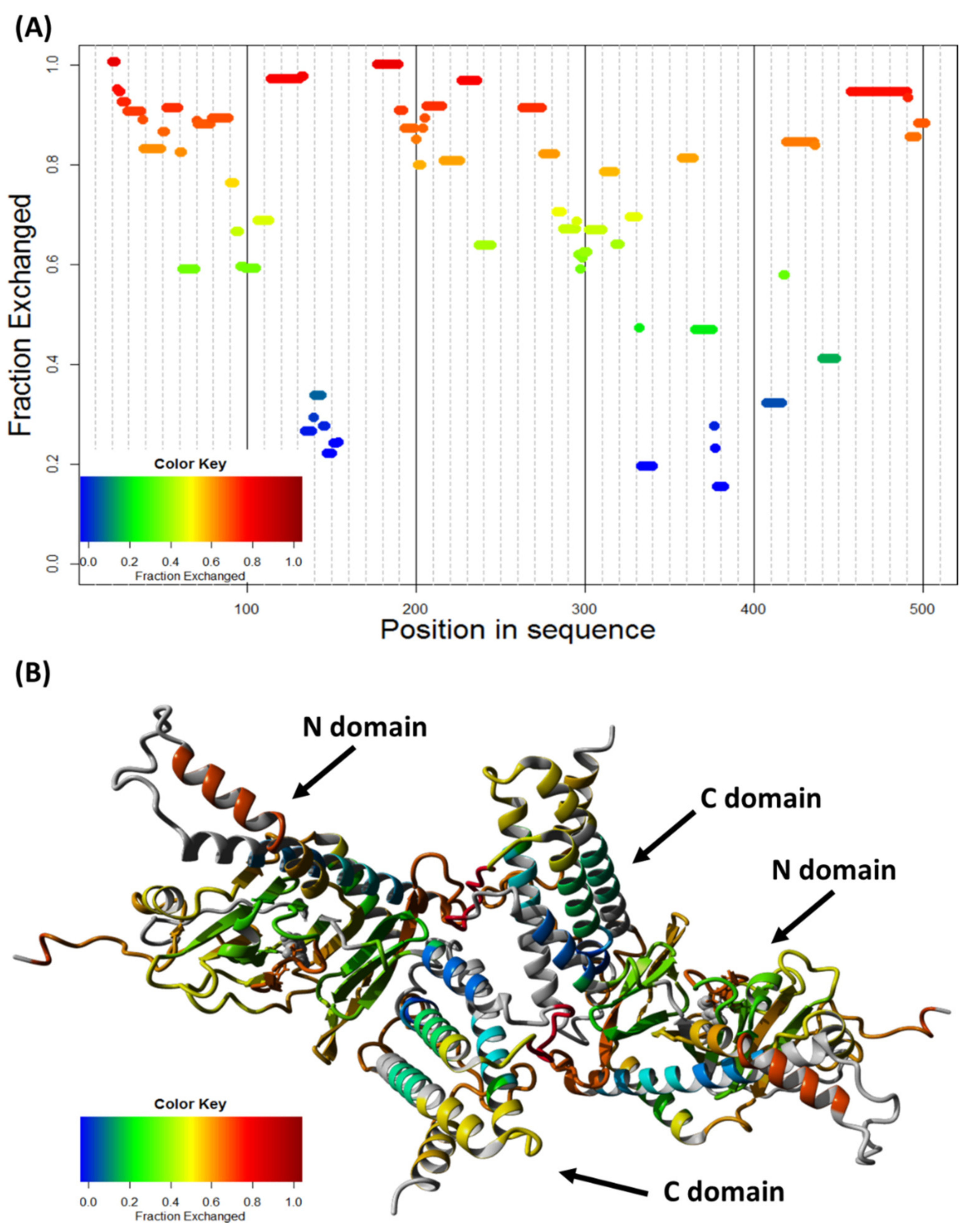
2.5. FTO Forms Homodimer via Its C-Terminal Domain
3. Discussion
4. Materials and Methods
4.1. cDNA Cloning and Plasmid Constructs
4.2. Site Directed Mutagenesis
4.3. Expression and Purification of FTO in E. coli
4.4. Expression and Purification of Proteins in Baculovirus Expression System (BES)
4.5. Preparation of Protein Samples
4.6. Molecular Modeling
4.7. Enzymatic Assay
4.8. Screening for Ligand Binding
4.8.1. Differential Scanning Fluorimetry
4.8.2. Label-Free nanoDSF
4.9. Microscale Thermophoresis (MST)
4.10. In Silico Analysis of FTO Amino Acid Sequence
4.11. Identification of FTO Phosphorylation Sites
4.12. FTO Dephosphorylation
4.13. Hydrogen–Deuterium Exchange (HDX)
4.14. Gel Filtration Chromatography
4.15. Small-Angle X-ray Scattering (SAXS)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-OG | 2-oxoglutarate |
| ACN | Acetonitrile |
| ALKBH | AlkB homolog |
| BES | Baculovirus Expression System |
| BMI | Body mass index |
| CaM | Calmodulin |
| DSF | Differential Scanning Fluorimetry |
| KD. | Dissociation constant |
| FTO | FaT mass and Obesity-associated |
| BESFTO | FTO protein expressed in baculovirus expression system |
| ECFTO | FTO protein expressed in E. coli |
| HDX-MS | Hydrogen–deuterium exchange monitored with MS |
| IRX3 | Iroquois-class homeodomain protein |
| LC-MS-MS/MS | Liquid chromatography coupled with tandem mass spectrometry |
| nanoDSF | Low-volume Differential Scanning Fluorimetry |
| MST | Microscale Thermophoresis |
| N6-meAm | N6,2′-O-dimethyladenosine |
| N6-meA | N6-methyladenosine |
| SNPs | Single Nucleotide Polymorphisms |
| SDM | Site directed mutagenesis |
| SAXS | Small-angle X-ray scattering |
| SFPQ | Splicing factor proline and glutamine rich |
| TFA | Trifluoroacetic acid |
References
- Groop, L. From fused toes in mice to human obesity. Nat. Genet. 2007, 39, 706–707. [Google Scholar] [CrossRef]
- Dina, C.; Meyre, D.; Gallina, S.; Durand, E.; Körner, A.; Jacobson, P.; Carlsson, L.M.S.; Kiess, W.; Vatin, V.; Lecoeur, C.; et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 2007, 39, 724–726. [Google Scholar] [CrossRef]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.B.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 5826, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauer, J.; Jaffrey, S.R. FTO, m6Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett. 2018, 592, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smemo, S.; Tena, J.J.; Kim, K.H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 7492, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Martin Carli, J.F.; LeDuc, C.A.; Zhang, Y.; Stratigopoulos, G.; Leibel, R.L. FTO mediates cell-autonomous effects on adipogenesis and adipocyte lipid content by regulating gene expression via 6mA DNA modifications 1. J. Lipid Res. 2018, 59, 1446–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachse, G.; Church, C.; Stewart, M.; Cater, H.; Teboul, L.; Cox, R.D.; Ashcroft, F.M. FTO demethylase activity is essential for normal bone growth and bone mineralization in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 843–850. [Google Scholar] [CrossRef]
- Cao, Y.; Zhuang, Y.; Chen, J.; Xu, W.; Shou, Y.; Huang, X.; Shu, Q.; Li, X. Dynamic effects of Fto in regulating the proliferation and differentiation of adult neural stem cells of mice. Hum. Mol. Genet. 2020, 29, 727–735. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Adamiak, M.; Mayourian, J.; Sassi, Y.; Liang, Y.; Agarwal, N.; Jha, D.; Zhang, S.; Kohlbrenner, E.; Chepurko, E.; et al. FTO-Dependent N6-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation 2018, 139, 518–532. [Google Scholar] [CrossRef]
- Hirayama, M.; Wei, F.Y.; Chujo, T.; Oki, S.; Yakita, M.; Kobayashi, D.; Araki, N.; Takahashi, N.; Yoshida, R.; Nakayama, H.; et al. FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep. 2020, 31, 107464. [Google Scholar] [CrossRef]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.G.; Sun, G.G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Flavell, R.A.; Li, H.B. RNA m6A modification and its function in diseases. Front. Med. 2018, 12, 481489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.; Dang, Y.; Chen, G.; Mo, Z. Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 2015, 8, 13405–13410. [Google Scholar] [PubMed]
- Xu, D.; Shao, W.; Jiang, Y.; Wang, X.; Liu, Y.; Liu, X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol. Rep. 2017, 38, 2285–2292. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Pilžys, T.; Marcinkowski, M.; Kukwa, W.; Garbicz, D.; Dylewska, M.; Ferenc, K.; Mieczkowski, A.; Kukwa, A.; Migacz, E.; Wołosz, D.; et al. ALKBH overexpression in head and neck cancer: Potential target for novel anticancer therapy. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Boissel, S.; Reish, O.; Proulx, K.; Kawagoe-Takaki, H.; Sedgwick, B.; Yeo, G.S.H.; Meyre, D.; Golzio, C.; Molinari, F.; Kadhom, N.; et al. Loss-of-Function Mutation in the Dioxygenase-Encoding FTO Gene Causes Severe Growth Retardation and Multiple Malformations. Am. J. Hum. Genet. 2009, 85, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Niu, T.; Chang, J.; Lei, X.; Zhao, M.; Wang, Q.; Cheng, W.; Wang, J.; Feng, Y.; Chai, J. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 2010, 464, 1205–1209. [Google Scholar] [CrossRef]
- Toh, J.D.W.; Sun, L.; Lau, L.Z.M.; Tan, J.; Low, J.J.A.; Tang, C.W.Q.; Cheong, E.J.Y.; Tan, M.J.H.; Chen, Y.; Hong, W.; et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N6-methyladenosine demethylase FTO. Chem. Sci. 2015, 6, 112–122. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 7637, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, C.; Lee, S.; Bagg, E.A.; McTaggart, J.S.; Deacon, R.; Gerken, T.; Lee, A.; Moir, L.; Mecinovic, J.; Quwailid, M.M.; et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet 2009, 5, e1000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosovic, M.; Molares, H.C.; Gregorova, P.; Hrossova, D.; Kudla, G.; Vanacova, S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res. 2017, 45, 11356–11370. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowski, M.; Pilžys, T.; Garbicz, D.; Steciuk, J.; Zugaj, D.; Mielecki, D.; Sarnowski, T.J.; Grzesiuk, E. Human and Arabidopsis alpha-ketoglutarate-dependent dioxygenase homolog proteins—New players in important regulatory processes. IUBMB Life 2020, 72, 1126–1144. [Google Scholar] [CrossRef]
- Tai, H.; Wang, X.; Zhou, J.; Han, X.; Fang, T.; Gong, H.; Huang, N.; Chen, H.; Qin, J.; Yang, M.; et al. Protein kinase Cβ activates fat mass and obesity-associated protein by influencing its ubiquitin/proteasome degradation. FASEB J. 2017, 31, 4396–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulds, K.J.; Egelston, J.N.; Sedivy, L.J.; Mitchell, M.K.; Garimella, S.; Kozlowski, H.; D’Alessandro, A.; Hansen, K.C.; Balsbaugh, J.L.; Phiel, C.J. Glycogen synthase kinase-3 (gsk-3) activity regulates mRNA methylation in mouse embryonic stem cells. J. Biol. Chem. 2018, 293, 10731–10743. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Wang, Y.; Wang, R.; Zhang, X.; Liu, Y.; Jia, G.; Chen, P.R. SFPQ Is an FTO-Binding Protein that Facilitates the Demethylation Substrate Preference. Cell Chem. Biol. 2020, 27, 283–291. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Woodbury, R.L. Complex behavior in solution of homodimeric SecA. Protein Sci. 2002, 11, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.-G.G.Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.G.Y.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, X.; Li, L.; Liu, Z.; Wang, Z. Use of baculovirus expression system for generation of virus-like particles: Successes and challenges. Protein Expr. Purif. 2013, 90, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Galperin, M.Y. The DxDxDG motif for calcium binding: Multiple structural contexts and implications for evolution. J. Mol. Biol. 2004, 343, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Santamaría-Hernando, S.; Krell, T.; Ramos-González, M.I. Identification of a novel calcium binding motif based on the detection of sequence insertions in the animal peroxidase domain of bacterial proteins. PLoS ONE 2012, 7, e40698. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 2015, 79, 28–29. [Google Scholar] [CrossRef]
- Giladi, M.; Khananshvili, D. Hydrogen-Deuterium Exchange Mass-Spectrometry of Secondary Active Transporters: From Structural Dynamics to Molecular Mechanisms. Front. Pharmacol. 2020, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; Das, A.; Chakrabarti, S.; Chakrabarti, O. Calcium dependent regulation of protein ubiquitination—Interplay between E3 ligases and calcium binding proteins. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1227–1235. [Google Scholar] [CrossRef]
- Guillou, J.L.; Nakata, H.; Cooper, D.M.F. Inhibition by calcium of mammalian adenylyl cyclases. J. Biol. Chem. 1999, 1864, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Lancaster, D.E.; McNeill, L.A.; McDonough, M.A.; Aplin, R.T.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem. J. 2004, 383, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 2006, 100, 64–669. [Google Scholar] [CrossRef] [PubMed]
- Waheed, S.O.; Ramanan, R.; Chaturvedi, S.S.; Ainsley, J.; Evison, M.; Ames, J.M.; Schofield, C.J.; Christov, C.Z.; Karabencheva-Christova, T.G. Conformational flexibility influences structure-function relationships in nucleic acid N-methyl demethylases. Org. Biomol. Chem. 2019, 17, 2223–2231. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Shelenkov, A.A.; Dunbrack, R.L. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003, 12, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-G.; Yi, C.; Duguid, E.M.; Sullivan, C.T.; Jian, X.; Rice, P.A.; He, C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 2008, 452, 961–965. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.; Jia, G.; Hou, G.; Dai, Q.; Zhang, W.; Zheng, G.; Jian, X.; Yang, C.-G.; Cui, Q.; He, C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature 2010, 468, 330–333. [Google Scholar] [CrossRef]
- Yi, C.; Chen, B.; Qi, B.; Zhang, W.; Jia, G.; Zhang, L.; Li, C.J.; Dinner, A.R.; Yang, C.-G.; He, C. Duplex interrogation by a direct DNA repair protein in search of base damage. Nat. Struct. Mol. Biol. 2012, 19, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Ching, H.Y.V.; Mascali, F.C.; Bertrand, H.C.; Bruch, E.M.; Demay-Drouhard, P.; Rasia, R.M.; Policar, C.; Tabares, L.C.; Un, S. The Use of Mn(II) Bound to His-tags as Genetically Encodable Spin-Label for Nanometric Distance Determination in Proteins. J. Phys. Chem. Lett. 2016, 7, 1072–1076. [Google Scholar] [CrossRef]
- Ramsey, J.; Ripley, B. pspline: Penalized Smoothing Splines. Available online: https://cran.r-project.org/package=pspline (accessed on 12 June 2019).
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.R-project.org (accessed on 12 June 2019).
- Bartoschik, T.; Galinec, S.; Kleusch, C.; Walkiewicz, K.; Breitsprecher, D.; Weigert, S.; Muller, Y.A.; You, C.; Piehler, J.; Vercruysse, T.; et al. Near-native, site-specific and purification-free protein labeling for quantitative protein interaction analysis by MicroScale Thermophoresis. Sci. Rep. 2018, 8, 4977. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Ferreira, I.; Bento, I.; Pimenta-Marques, A.; Jana, S.C.; Lince-Faria, M.; Duarte, P.; Borrego-Pinto, J.; Gilberto, S.; Amado, T.; Brito, D.; et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr. Biol. 2013, 23, 2245–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, G.; Pérez, J. Combined sampler robot and high-performance liquid chromatography: A fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009, 42, 892–900. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0 : Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
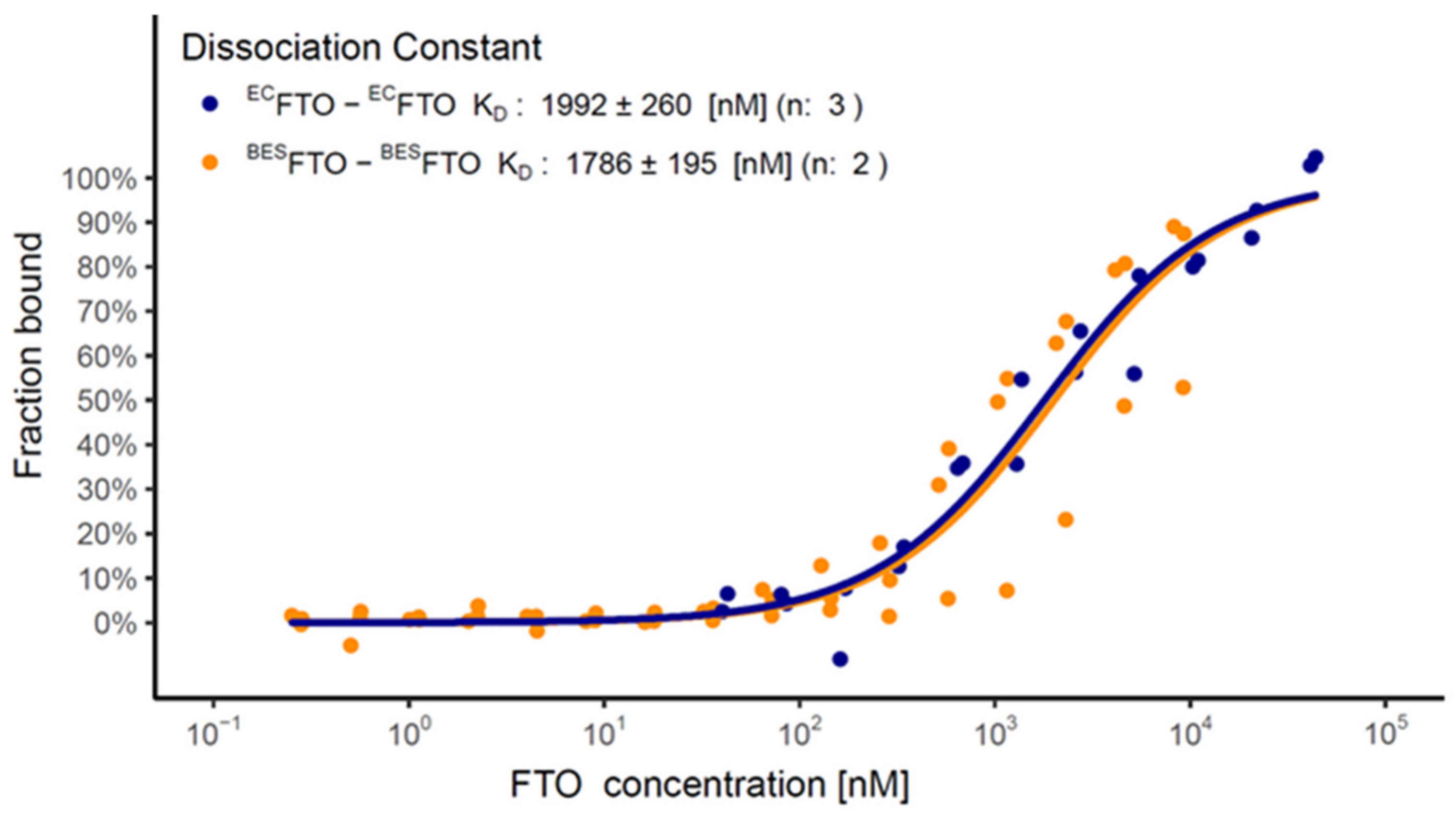{kind=link}
| Technique | Experiment | Buffer Composition | |
|---|---|---|---|
| SDM1 | Site Directed Mutagenesis | Preparation of FTO plasmids carrying point mutations S246G and S256D | 50 mM Tris-HCl, pH 9.0, 5% (v/v) DMSO, 20 mM (NH4)2SO4, 2.5 mM MgCl2, 200 µM each dNTP, 0.5 µM each primer, 1 U of Marathon Polymerase (#1003-200, A&A Biotechnology), 0.8 ng of pET-28a(+) carrying hFTO cDNA |
| SOC | Bacteria culturing | Transformation | 10 mM NaCl, 2.5 mM KCl, 2% (w/v) tryptone, 0.5% (w/v) yeast extract, 20 mM glucose, 10 mM MgCl2, 10 mM MgSO4 |
| PRP1 | Protein purification | Lysis bacteria | 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 10% (v/v) glycerol, 0.1% (v/v) TWEEN® 20, 0.1% (w/v) lysozyme, 5 mM imidazole, 5 mM β-mercaptoethanol, |
| PRP2 | Protein purification | Elution from Ni-charged Resin | 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl, 10% (v/v) glycerol, 0.1% (v/v) TWEEN® 20, 150 mM imidazole, 5 mM β-mercaptoethanol, |
| PRP3 | Protein purification | Dialysis | 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 50% (v/v) glycerol, 5mM β-mercaptoethanol |
| PRP4 | Protein purification | Lysis of BES insect cells | 50 mM HEPES, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 5 mM β-mercaptoethanol |
| TSA1 | Differential Scanning Fluorimetry | Evaluation of apo BESFTO thermal stability | 50 mM HEPES, pH 7.5, 150 mM NaCl, 1% (v/v) SYPRO® ORANGE stock (#S6650, Life Technologies), 2 mM L-ascorbic acid |
| TSA2 | Differential Scanning Fluorimetry | Evaluation of effect of calcium on apo BESFTO thermal stability | 50 mM HEPES, pH 7.5, 150 mM NaCl, 1% (v/v) SYPRO® ORANGE stock, 2 mM L-ascorbic acid, 0.5 mM CaCl2 |
| TSA3 | Differential Scanning Fluorimetry | Evaluation of holo BESFTO thermal stability | 50 mM HEPES, pH 7.5, 150 mM NaCl, 1% (v/v) SYPRO® ORANGE stock, 2 mM L-ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2 |
| TSA4 | Differential Scanning Fluorimetry | Evaluation of effect of calcium on holo BESFTO thermal stability | 50 mM HEPES pH 7.5, 150 mM NaCl, 1% (v/v) SYPRO® ORANGE stock and 2 mM L-ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2, 0.5 mM CaCl2 |
| DSF1 | Nano Differential Scanning Fluorimetry | Evaluation of apo ECFTO and apo BESFTO thermal stability | 50 mM MES, pH 6.5, 150 mM NaCl, 10% (v/v) glycerol |
| DSF2 | Nano Differential Scanning Fluorimetry | Evaluation of effect of Mn2+ on BESFTO thermal stability | 50 mM MES, pH 6.5, 150 mM NaCl, 10% (v/v) glycerol, 0.5 mM MnCl2 |
| DSF3 | Nano Differential Scanning Fluorimetry | Evaluation effect of 2-OG on BESFTO thermal stability | 50 mM MES, pH 6.5, 150 mM NaCl, 10% (v/v) glycerol, 1mM 2-OG |
| DSF4 | Nano Differential Scanning Fluorimetry | Evaluation of holo ECFTO and holo BESFTO thermal stability | 50 mM MES, pH 6.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM 2-OG, 0.5 mM MnCl2, |
| DSF5 | Nano Differential Scanning Fluorimetry | Evaluation of effect of dephosphorylation on ECFTO and BESFTO thermal stability | 37 mM HEPES, 1 mM MES, pH 7.5, 124 mM NaCl, 37% (v/v) glycerol, 0.01% Brij 35, 3.7 mM β-mercaptoethanol, 2 mM DTT, 1 mM 2-OG, 1 mM Mn Cl2 |
| DSF6 | Nano Differential Scanning Fluorimetry | Evaluation effect of dephosphorylation on ECFTO and BESFTO thermal stability | 37 mM HEPES, 1mM MES, pH 7.5, 124 mM NaCl, 37% (v/v) glycerol, 0.01% Brij 35, 3.7 mM β-mercaptoethanol, 2 mM DTT, 1 mM 2-OG, 1 mM Mn Cl2, 800 U of Lambda Protein Phosphatase (#P0753S, New England BioLabs) |
| DEP1 | Dephosphorylation | Dephosphorylation of ECFTO and BESFTO | 37 mM HEPES, 1mM MES, pH 7.5, 150 mM NaCl, 37% (v/v) glycerol, 3.7 mM β-mercaptoethanol, 1 mM 2-OG, 1 mM MnCl2, 1x NEBuffer for Protein MetalloPhosphatases (#B0761S, New England BioLabs) |
| HDX1 | Hydrogen–Deuterium Exchange | Sample preincubation | 50 mM MES, pH 6.1, 150 mM NaCl, 0.004% (v/v) TWEEN® 20, 2 mM L-ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2 |
| HDX2 | Hydrogen–Deuterium Exchange | Sample preincubation with Ca2+ | 50 mM MES pH 6.1, 150 mM NaCl, 0.004% (v/v) TWEEN® 20, 2 mM L-ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2, 0.5 mM CaCl2 |
| HDX3 | Hydrogen–Deuterium Exchange | HDX reaction | 30 mM Tris-DCl, pD 7.5 (pHREAD+0.4), 150 mM NaCl in D2O (#DLM-4DR-99.8-PK, Cambridge Isotope Laboratories, Inc.) |
| HDX4 | Hydrogen–Deuterium Exchange | HDX stopping | 2 M glycine, pH 2.4, 107 mM NaCl |
| SEC1 | Gel Filtration Chromatography | Estimation of molecular size of protein | 50 mM MES, pH 6.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM 2-OG, 0.5 mM MnCl2 |
| MST1 | Microscale thermophoresis | Evaluation of dimerization and interaction with Fe2+ and 2-OG of BESFTO | 50 mM MES, pH 6.1, 150 mM NaCl, 0.05% (v/v) TWEEN® 20, 2 mM L-ascorbic acid |
| MST2 | Microscale thermophoresis | Evaluation of Fe2+ effect on dimerization and interaction with 2-OG of BESFTO | 50 mM MES, pH 6.1, 150 mM NaCl, 0.05% (v/v) TWEEN® 20, 2 mM L-ascorbic acid, 0.5 mM (NH4)2Fe(SO4)2 |
| MST3 | Microscale thermophoresis | Evaluation of 2-OG effect on dimerization and interaction with Fe2+ of BESFTO | 50 mM MES pH 6.1, 150 mM NaCl, 0.05% (v/v) TWEEN® 20, 2 mM L-ascorbic acid, 1 mM 2-OG |
| MST4 | Microscale thermophoresis | Evaluation of dimerization of ECFTO and BESFTO | 50 mM MES pH 6.1, 150 mM NaCl, 0.05% (v/v) TWEEN® 20, 2 mM L-ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2 |
| SAX1 | Small-angle X-ray Scattering | Evaluation of holo BESFTO structure in water solution | 33 mM HEPES, 13 mM MES, pH 7.0, 150 mM NaCl, 7.3% (v/v) glycerol, 3.3 mM β-mercaptoethanol, 2 mM ascorbic acid, 1 mM 2-OG, 0.5 mM (NH4)2Fe(SO4)2, 0.5 mM CaCl2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcinkowski, M.; Pilžys, T.; Garbicz, D.; Piwowarski, J.; Mielecki, D.; Nowaczyk, G.; Taube, M.; Gielnik, M.; Kozak, M.; Winiewska-Szajewska, M.; et al. Effect of Posttranslational Modifications on the Structure and Activity of FTO Demethylase. Int. J. Mol. Sci. 2021, 22, 4512. https://doi.org/10.3390/ijms22094512
Marcinkowski M, Pilžys T, Garbicz D, Piwowarski J, Mielecki D, Nowaczyk G, Taube M, Gielnik M, Kozak M, Winiewska-Szajewska M, et al. Effect of Posttranslational Modifications on the Structure and Activity of FTO Demethylase. International Journal of Molecular Sciences. 2021; 22(9):4512. https://doi.org/10.3390/ijms22094512
Chicago/Turabian StyleMarcinkowski, Michał, Tomaš Pilžys, Damian Garbicz, Jan Piwowarski, Damian Mielecki, Grzegorz Nowaczyk, Michał Taube, Maciej Gielnik, Maciej Kozak, Maria Winiewska-Szajewska, and et al. 2021. "Effect of Posttranslational Modifications on the Structure and Activity of FTO Demethylase" International Journal of Molecular Sciences 22, no. 9: 4512. https://doi.org/10.3390/ijms22094512
APA StyleMarcinkowski, M., Pilžys, T., Garbicz, D., Piwowarski, J., Mielecki, D., Nowaczyk, G., Taube, M., Gielnik, M., Kozak, M., Winiewska-Szajewska, M., Szołajska, E., Dębski, J., Maciejewska, A. M., Przygońska, K., Ferenc, K., Grzesiuk, E., & Poznański, J. (2021). Effect of Posttranslational Modifications on the Structure and Activity of FTO Demethylase. International Journal of Molecular Sciences, 22(9), 4512. https://doi.org/10.3390/ijms22094512