
Role of PGC-1α in the Mitochondrial NAD+ Pool in Metabolic Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
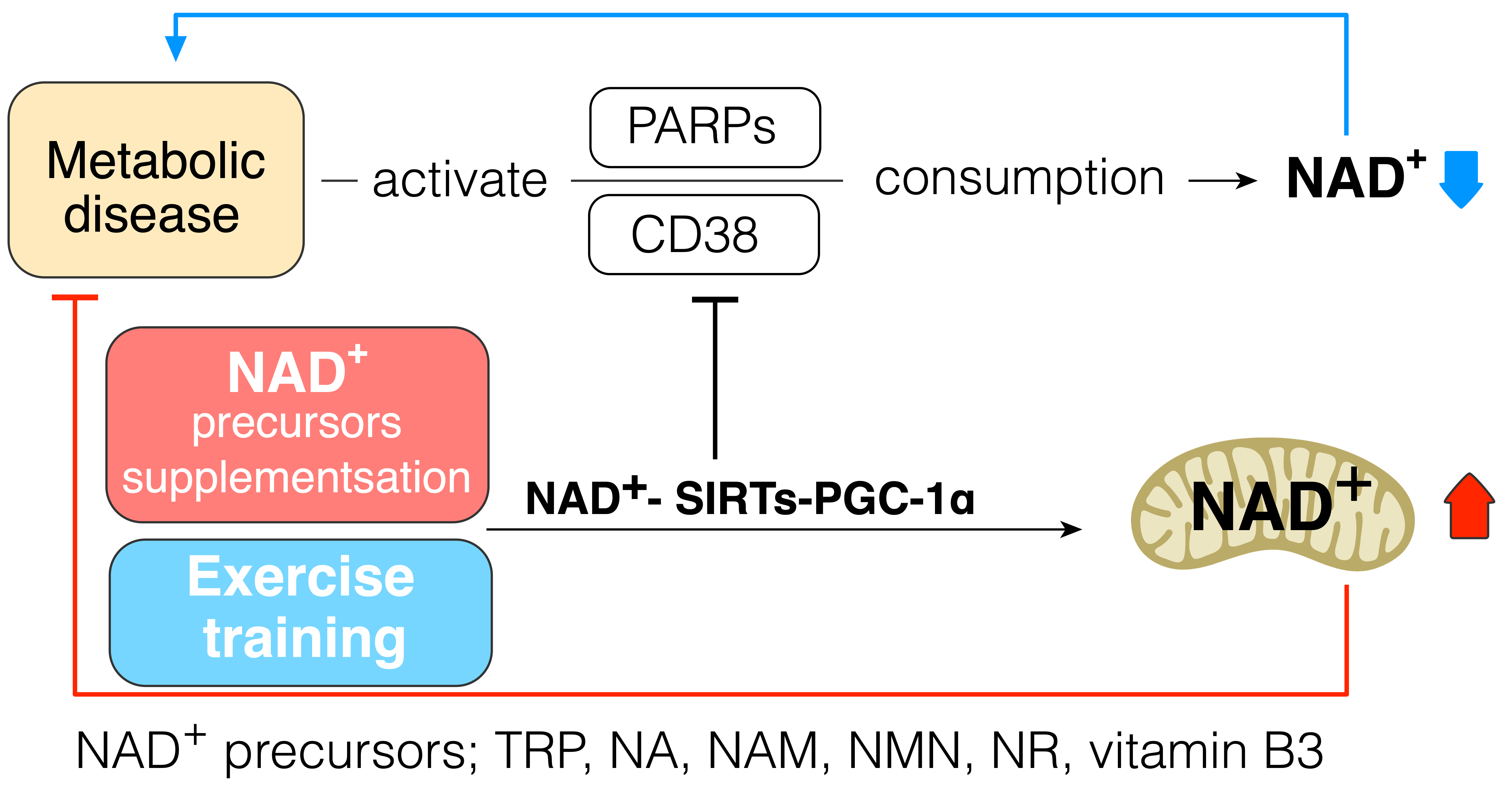
2. NAD+–SIRT1–PGC-1α Pathway in Metabolic Diseases
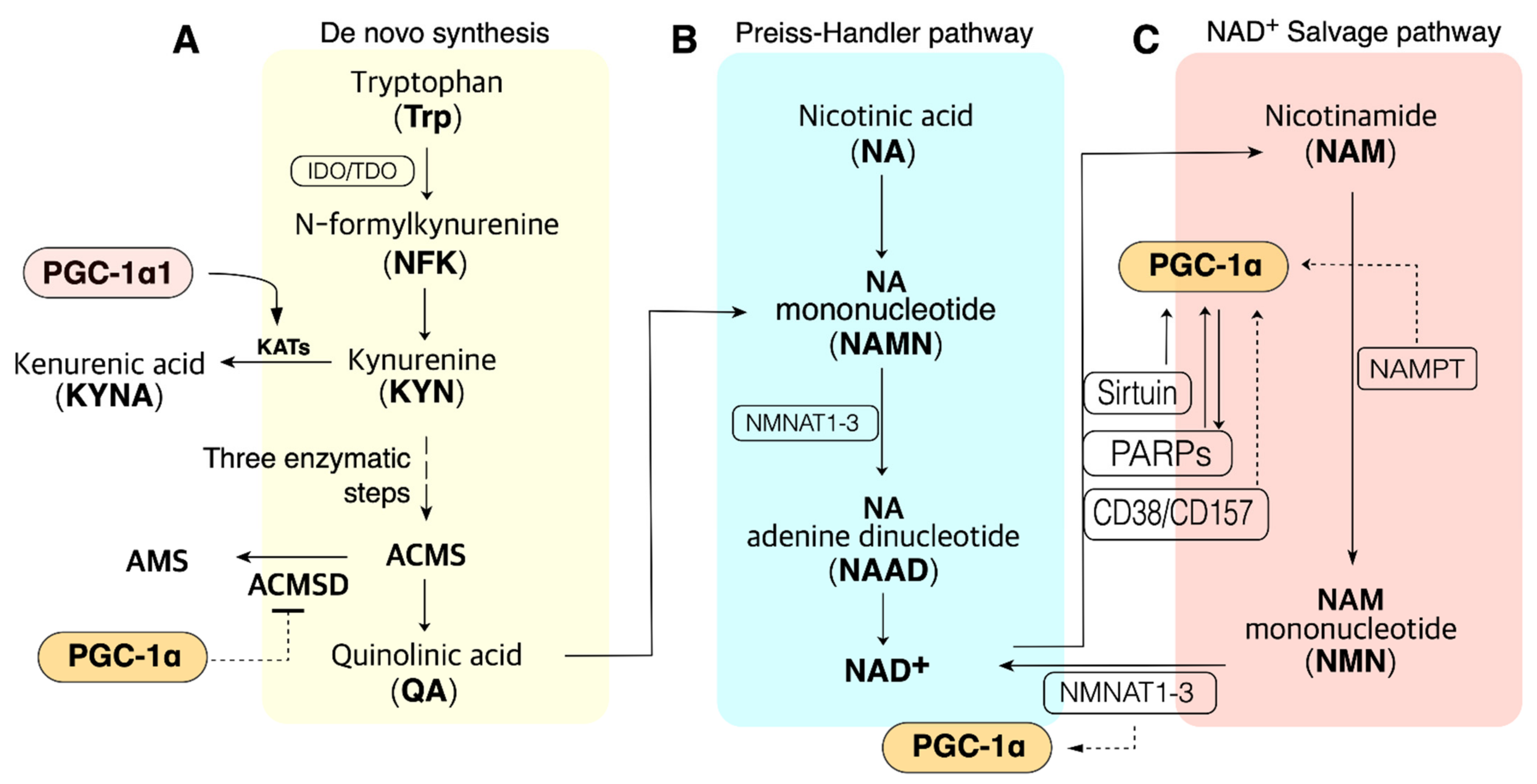
3. NAD+ Biosynthesis
3.1. The Salvage Pathway
3.2. The Preiss–Handler Pathway
3.3. The De Novo Synthesis (DNS) Pathway
3.4. NAD+ Consumption as a Cosubstrate
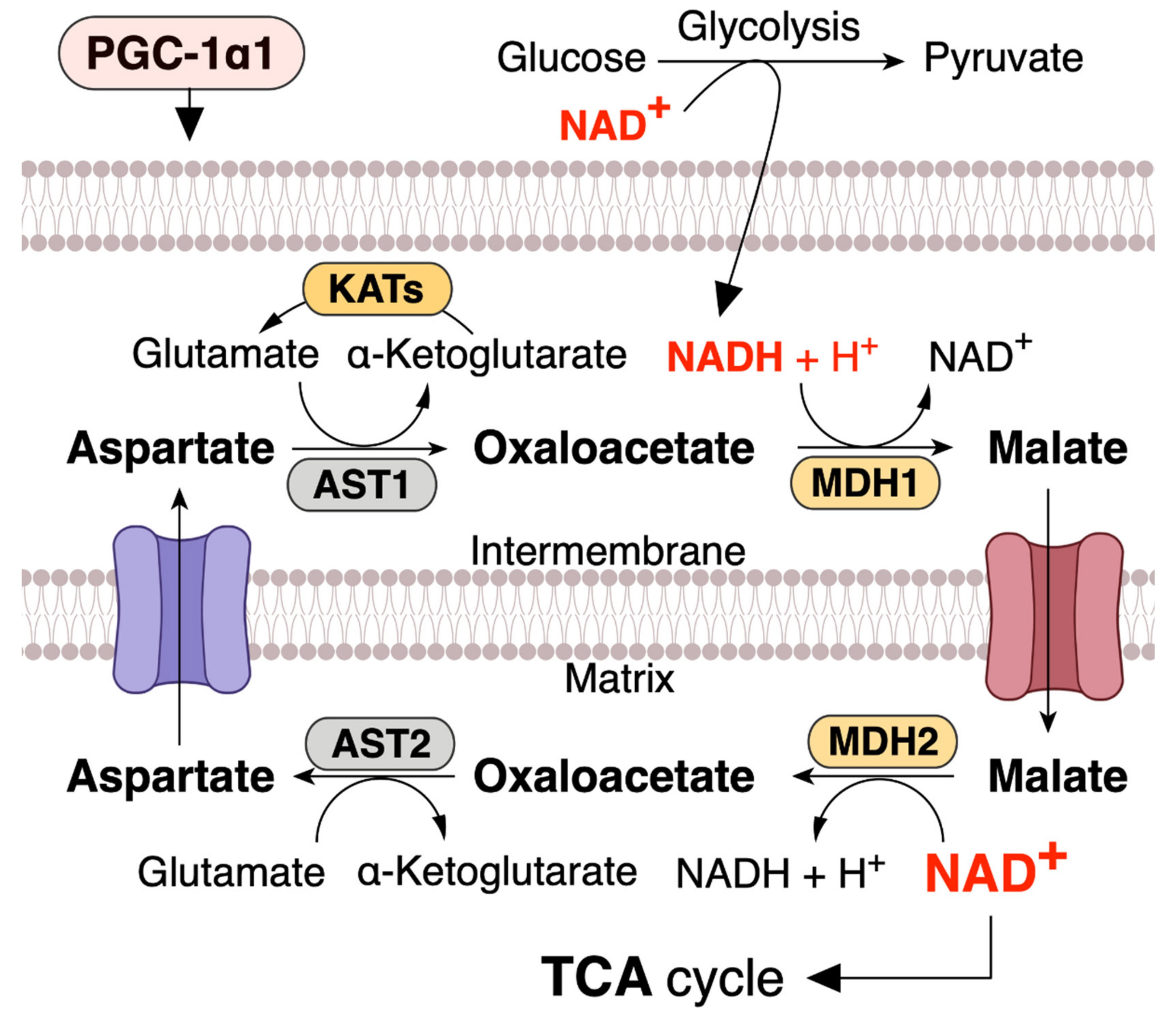
4. PGC-1α1 Regulates the Mitochondrial NAD+ Pool via Malate–Aspartate Shuttle
5. Role of PGC-1α in NAD+ Metabolism in Metabolic Diseases
5.1. NAMPT–PGC-1α
5.2. PARP1–PGC-1α
5.3. CD38–PGC-1α
5.4. ACMSD
5.5. NNMT
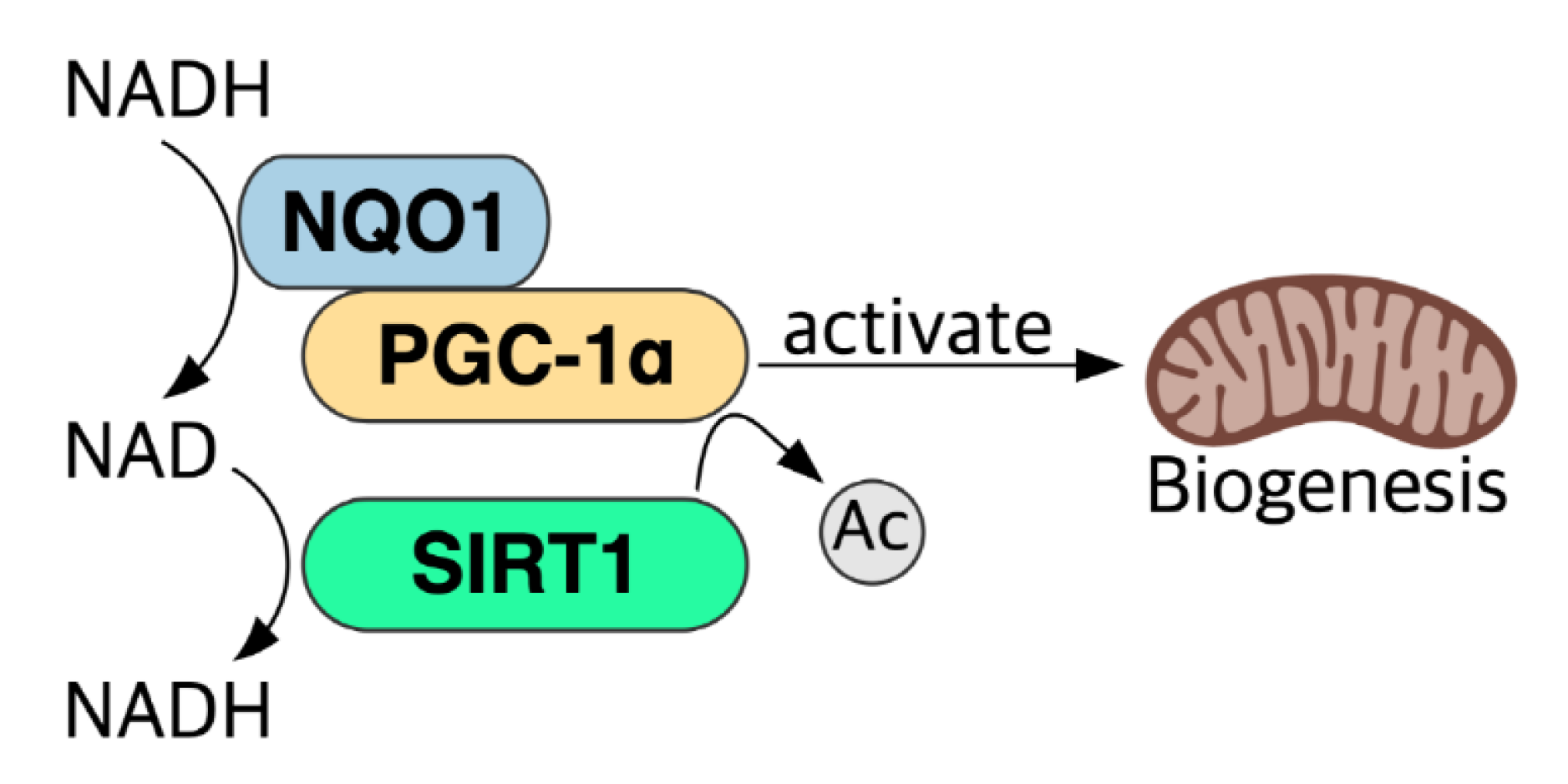
5.6. NADH–NQO1–PGC-1α Pathway and NAD+ Level
6. NAD+–SIRT1–PGC-1α Pathway in Diabetes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 89, 1946–1955. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 535–546. [Google Scholar] [CrossRef]
- Johnson, S.; Imai, S.I. NAD (+) biosynthesis, aging, and disease. F1000Res 2018, 7, 132. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902. [Google Scholar] [CrossRef] [Green Version]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.Y.; Russell, J.W. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int. Rev. Neurobiol. 2019, 145, 177–209. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The protein level of PGC-1α, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, J.M.; Hindle, A. Living in a box or call of the wild? Revisiting lifetime inactivity and sarcopenia. Antioxid. Redox Signal. 2011, 15, 2529–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Guia, R.M.; Agerholm, M.; Nielsen, T.S.; Consitt, L.A.; Sogaard, D.; Helge, J.W.; Larsen, S.; Brandauer, J.; Houmard, J.A.; Treebak, J.T. Aerobic and resistance exercise training reverses age-dependent decline in NAD(+) salvage capacity in human skeletal muscle. Physiol. Rep. 2019, 7, e14139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Zorzano, A.; Hernandez-Alvarez, M.I.; Palacin, M.; Mingrone, G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1alpha or PGC-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1028–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Kume, S.; Kanasaki, K.; Takeda-Watanabe, A.; Koya, D. Sirtuins as possible drug targets in type 2 diabetes. Curr. Drug Targets 2013, 14, 622–636. [Google Scholar] [CrossRef]
- Garcia Soriano, F.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Peterson, B.S.; Bomze, H.M.; Hirschey, M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metab. 2015, 26, 486–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, J.; Chen, L.; Tang, Q.; Zhang, J.; Li, Y.; He, J. The Role of Sirt6 in Obesity and Diabetes. Front. Physiol. 2018, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT activation can contribute to the development of fatty liver disease by modulating the NAD (+) metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1alpha signalling pathway. Adipocyte 2020, 9, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, R.Y.; Song, J.; Guan, Y.F.; Xu, T.Y.; Du, H.; Viollet, B.; Miao, C.Y. Loss of AMP-activated protein kinase-alpha2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes 2012, 61, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Waldman, M.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Kornwoski, R.; Aravot, D.; Arad, M.; Hochhauser, E. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1α axis. Exp. Cell Res. 2018, 373, 112–118. [Google Scholar] [CrossRef] [PubMed]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: Potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [Green Version]
- Fröjdö, S.; Durand, C.; Molin, L.; Carey, A.L.; El-Osta, A.; Kingwell, B.A.; Febbraio, M.A.; Solari, F.; Vidal, H.; Pirola, L. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol. Cell Endocrinol. 2011, 335, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005, 2, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Ma, X.J.; Wu, L.N.; Zhao, Y.Y.; Zhang, P.Y.; Zhang, Y.H.; Shao, M.W.; Liu, F.; Li, F.; Qin, G.J. SIRT1 attenuates high glucose-induced insulin resistance via reducing mitochondrial dysfunction in skeletal muscle cells. Exp. Biol. Med. (Maywood) 2015, 240, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L.; Franklin, H. Epstein Lecture: Sirtuins, aging, and medicine. N. Engl. J. Med. 2011, 364, 2235–2244. [Google Scholar] [CrossRef]
- Imai, S. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 2009, 15, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, F.; Hirrlinger, J. The NAD+ /NADH redox state in astrocytes: Independent control of the NAD+ and NADH content. J. Neurosci. Res. 2011, 89, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Andris, F.; Van Gool, F.; Leo, O. Reconstructing eukaryotic NAD metabolism. Bioessays 2003, 25, 683–690. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Huang, R.X.; Tao, J. Nicotinamide mononucleotide attenuates glucocorticoid‑induced osteogenic inhibition by regulating the SIRT1/PGC‑1α signaling pathway. Mol. Med. Rep. 2020, 22, 145–154. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Enzymatic synthesis of nicotinamide mononucleotide. J. Biol. Chem. 1957, 225, 759–770. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef] [Green Version]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mázala, D.A.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Balducci, E.; Emanuelli, M.; Raffaelli, N.; Ruggieri, S.; Amici, A.; Magni, G.; Orsomando, G.; Polzonetti, V.; Natalini, P. Assay methods for nicotinamide mononucleotide adenylyltransferase of wide applicability. Anal. Biochem. 1995, 228, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Song, E.K.; Rah, S.Y.; Lee, Y.R.; Yoo, C.H.; Kim, Y.R.; Yeom, J.H.; Park, K.H.; Kim, J.S.; Kim, U.H.; Han, M.K. Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J. Biol. Chem. 2011, 286, 44480–44490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Krüger, A.; Bürkle, A.; Hauser, K.; Mangerich, A. Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy. Nat. Commun. 2020, 11, 2174. [Google Scholar] [CrossRef]
- Zhang, T.; Berrocal, J.G.; Yao, J.; DuMond, M.E.; Krishnakumar, R.; Ruhl, D.D.; Ryu, K.W.; Gamble, M.J.; Kraus, W.L. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J. Biol. Chem. 2012, 287, 12405–12416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria, bioenergetics, and the epigenome in eukaryotic and human evolution. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharmacol. 2006, 71, 399–407. [Google Scholar] [CrossRef]
- Agudelo, L.Z.; Ferreira, D.M.S.; Dadvar, S.; Cervenka, I.; Ketscher, L.; Izadi, M.; Zhengye, L.; Furrer, R.; Handschin, C.; Venckunas, T.; et al. Skeletal muscle PGC-1α1 reroutes kynurenine metabolism to increase energy efficiency and fatigue-resistance. Nat. Commun. 2019, 10, 2767. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [Green Version]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD⁺. Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef] [Green Version]
- Easlon, E.; Tsang, F.; Skinner, C.; Wang, C.; Lin, S.J. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008, 22, 931–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhouse, W.V.; Lehninger, A.L. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 1976, 36, 1392–1396. [Google Scholar]
- del Arco, A.; Morcillo, J.; Martínez-Morales, J.R.; Galián, C.; Martos, V.; Bovolenta, P.; Satrústegui, J. Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin during development and in adult rat tissues. Eur. J. Biochem. 2002, 269, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005, 79, 216–223. [Google Scholar] [CrossRef]
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007, 85, 3378–3385. [Google Scholar] [CrossRef] [PubMed]
- Pittelli, M.; Formentini, L.; Faraco, G.; Lapucci, A.; Rapizzi, E.; Cialdai, F.; Romano, G.; Moneti, G.; Moroni, F.; Chiarugi, A. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 2010, 285, 34106–34114. [Google Scholar] [CrossRef] [Green Version]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeks, M.H.; Shamseldin, H.E.; Alhashem, A.; Hashem, M.; Abdulwahab, F.; Alshedi, T.; Alobaid, I.; Zwartkruis, F.; Westland, D.; Fuchs, S.; et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum. Genet. 2019, 138, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- White, A.T.; Schenk, S. NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E308–E321. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xu, M.; Lee, J.; He, C.; Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1234–E1244. [Google Scholar] [CrossRef] [Green Version]
- Costford, S.R.; Bajpeyi, S.; Pasarica, M.; Albarado, D.C.; Thomas, S.C.; Xie, H.; Church, T.S.; Jubrias, S.A.; Conley, K.E.; Smith, S.R. Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E117–E126. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, S.; Arimoto, A.; Kuramoto, Y.; Kozako, T.; Honda, S.; Shimeno, H.; Soeda, S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol. Cell Biochem. 2010, 339, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinnler, R.; Gorski, T.; Stolz, K.; Schuster, S.; Garten, A.; Beck-Sickinger, A.G.; Engelse, M.A.; de Koning, E.J.; Körner, A.; Kiess, W.; et al. The adipocytokine Nampt and its product NMN have no effect on beta-cell survival but potentiate glucose stimulated insulin secretion. PLoS ONE 2013, 8, e54106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Wan, Z.; Root-McCaig, J.; Castellani, L.; Kemp, B.E.; Steinberg, G.R.; Wright, D.C. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring) 2014, 22, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, X.; Yang, H.; Xu, E.; Wu, S.; Wei, W.; Chen, J. Analysis of poly(ADP-ribose) polymerase-1 by enzyme-initiated auto-PARylation-controlled aggregation of hemin-graphene nanocomposites. Analyst 2018, 143, 2501–2507. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Lloret, A.; Beal, M.F. PGC-1α, Sirtuins and PARPs in Huntington’s Disease and Other Neurodegenerative Conditions: NAD+ to Rule Them All. Neurochem. Res. 2019, 44, 2423–2434. [Google Scholar] [CrossRef]
- Masutani, M.; Suzuki, H.; Kamada, N.; Watanabe, M.; Ueda, O.; Nozaki, T.; Jishage, K.; Watanabe, T.; Sugimoto, T.; Nakagama, H.; et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 2301–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.H.; Kim, D.W.; Jo, E.J.; Kim, Y.K.; Jo, Y.S.; Park, J.H.; Yoo, S.K.; Park, M.K.; Kwak, T.H.; Kho, Y.L.; et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes 2009, 58, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, A.; Long, D.J., 2nd; Stringer, J.L.; Jaiswal, A.K. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol. Chem. 2001, 276, 22559–22564. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Park, A.H.; Lee, S.H.; Lee, S.H.; Kim, J.H.; Yang, S.J.; Yeom, Y.I.; Kwak, T.H.; Lee, D.; Lee, S.J.; et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS ONE 2012, 7, e47122. [Google Scholar] [CrossRef] [PubMed]
- Ross, D. Quinone reductases multitasking in the metabolic world. Drug Metab Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscovitz, O.; Tsvetkov, P.; Hazan, N.; Michaelevski, I.; Keisar, H.; Ben-Nissan, G.; Shaul, Y.; Sharon, M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol. Cell 2012, 47, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadka, D.; Kim, H.J.; Oh, G.S.; Shen, A.; Lee, S.; Lee, S.B.; Sharma, S.; Kim, S.Y.; Pandit, A.; Choe, S.K.; et al. Augmentation of NAD(+) levels by enzymatic action of NAD(P)H quinone oxidoreductase 1 attenuates adriamycin-induced cardiac dysfunction in mice. J. Mol. Cell Cardiol. 2018, 124, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Oh, G.S.; Shen, A.; Lee, S.B.; Choe, S.K.; Kwon, K.B.; Lee, S.; Seo, K.S.; Kwak, T.H.; Park, R.; et al. Augmentation of NAD(+) by NQO1 attenuates cisplatin-mediated hearing impairment. Cell Death Dis. 2014, 5, e1292. [Google Scholar] [CrossRef] [Green Version]
- Di Francesco, A.; Choi, Y.; Bernier, M.; Zhang, Y.; Diaz-Ruiz, A.; Aon, M.A.; Kalafut, K.; Ehrlich, M.R.; Murt, K.; Ali, A.; et al. NQO1 protects obese mice through improvements in glucose and lipid metabolism. NPJ Aging Mech. Dis. 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Meex, R.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 2010, 53, 1714–1721. [Google Scholar] [CrossRef] [Green Version]
- Trevellin, E.; Scorzeto, M.; Olivieri, M.; Granzotto, M.; Valerio, A.; Tedesco, L.; Fabris, R.; Serra, R.; Quarta, M.; Reggiani, C.; et al. Exercise Training Induces Mitochondrial Biogenesis and Glucose Uptake in Subcutaneous Adipose Tissue Through eNOS-Dependent Mechanisms. Diabetes 2014, 63, 2800. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Moreno-Santos, I.; Pérez-Belmonte, L.M.; Macías-González, M.; Mataró, M.J.; Castellano, D.; López-Garrido, M.; Porras-Martín, C.; Sánchez-Fernández, P.L.; Gómez-Doblas, J.J.; Cardona, F.; et al. Type 2 diabetes is associated with decreased PGC1α expression in epicardial adipose tissue of patients with coronary artery disease. J. Transl. Med. 2016, 14, 243. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.J.; Choi, J.M.; Kim, L.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; Park, S.W.; Park, C.Y. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. J. Nutr. Biochem. 2014, 25, 66–72. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- van de Weijer, T.; Phielix, E.; Bilet, L.; Williams, E.G.; Ropelle, E.R.; Bierwagen, A.; Livingstone, R.; Nowotny, P.; Sparks, L.M.; Paglialunga, S.; et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 2015, 64, 1193–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Xiang, D.; Zhang, Q.; Wang, Y.T. Effectiveness of niacin supplementation for patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Medicine (Baltimore) 2020, 99, e21235. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.H.; Hancock, C.R.; Han, D.H.; Holloszy, J.O.; Nair, K.S.; Dasari, S. AMPK and PPARβ positive feedback loop regulates endurance exercise training-mediated GLUT4 expression in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E931–E939. [Google Scholar] [CrossRef] [PubMed]
- Costford, S.R.; Brouwers, B.; Hopf, M.E.; Sparks, L.M.; Dispagna, M.; Gomes, A.P.; Cornnell, H.H.; Petucci, C.; Phelan, P.; Xie, H.; et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol. Metab. 2018, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koh, J.-H.; Kim, J.-Y. Role of PGC-1α in the Mitochondrial NAD+ Pool in Metabolic Diseases. Int. J. Mol. Sci. 2021, 22, 4558. https://doi.org/10.3390/ijms22094558
Koh J-H, Kim J-Y. Role of PGC-1α in the Mitochondrial NAD+ Pool in Metabolic Diseases. International Journal of Molecular Sciences. 2021; 22(9):4558. https://doi.org/10.3390/ijms22094558
Chicago/Turabian StyleKoh, Jin-Ho, and Jong-Yeon Kim. 2021. "Role of PGC-1α in the Mitochondrial NAD+ Pool in Metabolic Diseases" International Journal of Molecular Sciences 22, no. 9: 4558. https://doi.org/10.3390/ijms22094558
APA StyleKoh, J. -H., & Kim, J. -Y. (2021). Role of PGC-1α in the Mitochondrial NAD+ Pool in Metabolic Diseases. International Journal of Molecular Sciences, 22(9), 4558. https://doi.org/10.3390/ijms22094558