
Caenorhabditis elegans as a Model System for Duchenne Muscular Dystrophy



Abstract
:1. Introduction
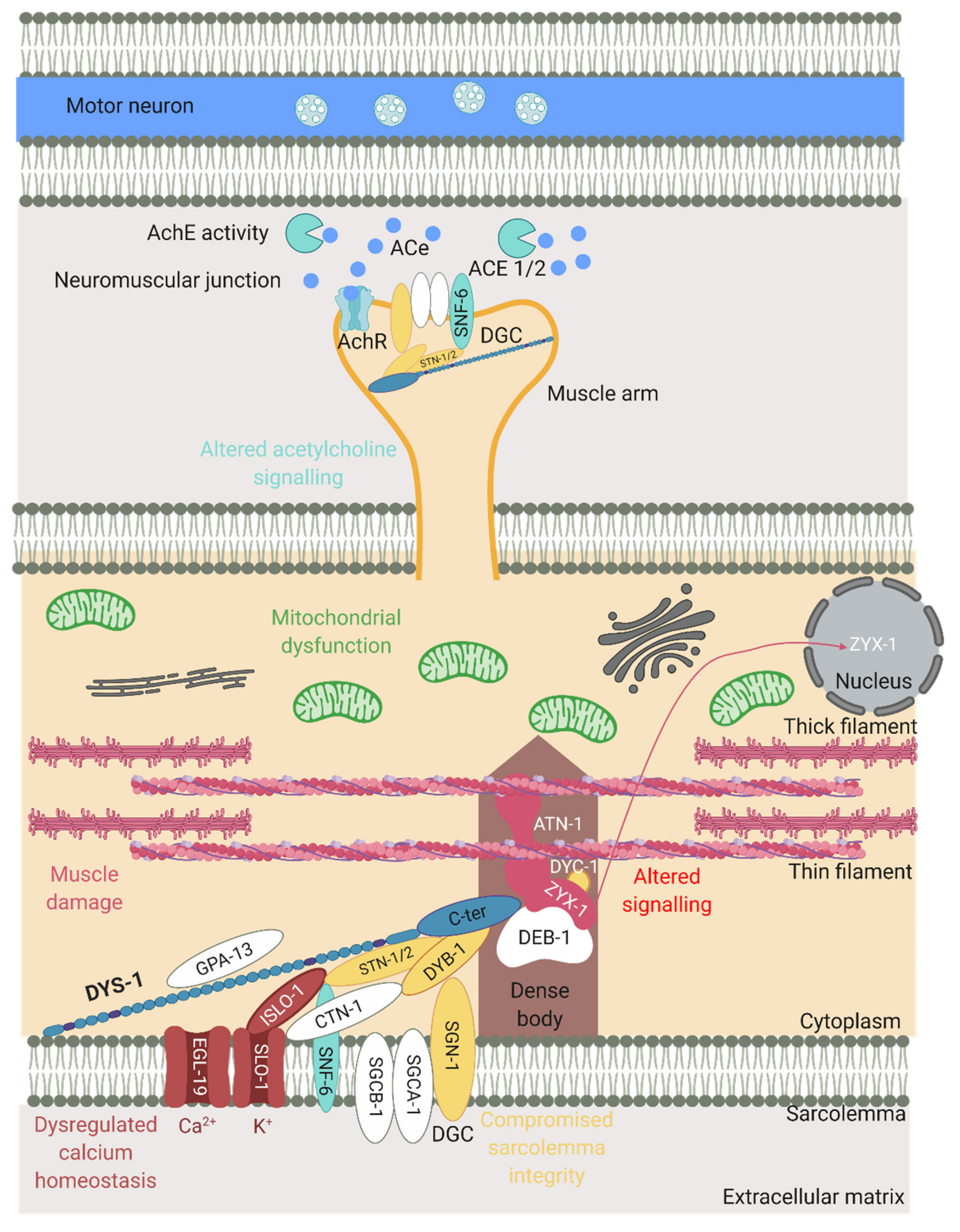
2. Phenotypes Observed in C. elegans DMD Mutants
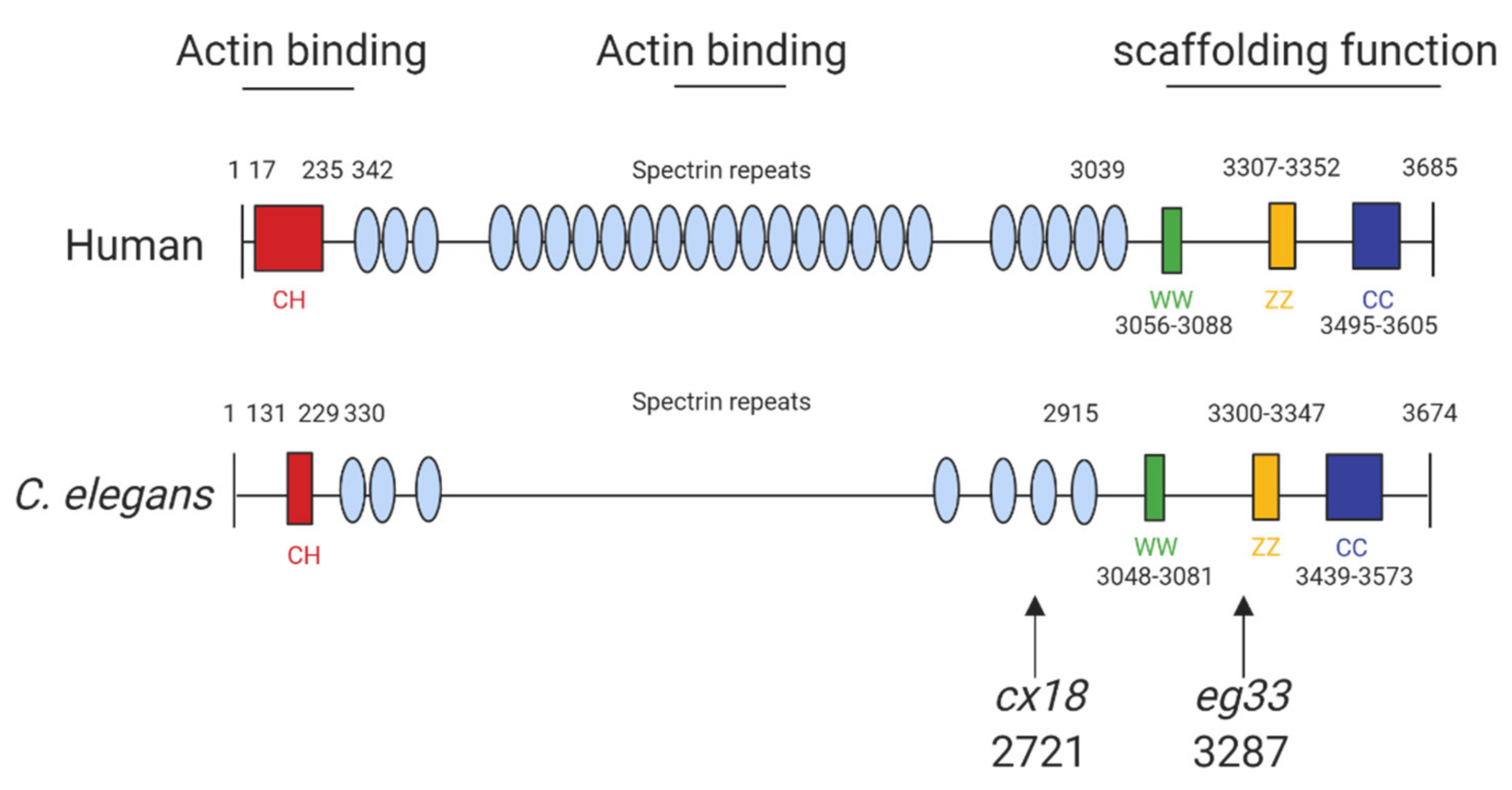
2.1. The dys-1 Single Mutant
2.2. Enhancing the Phenotype of dys-1 Mutants
2.3. Novel Mutation in dys-1
3. Effects of dys-1 on Gene Expression
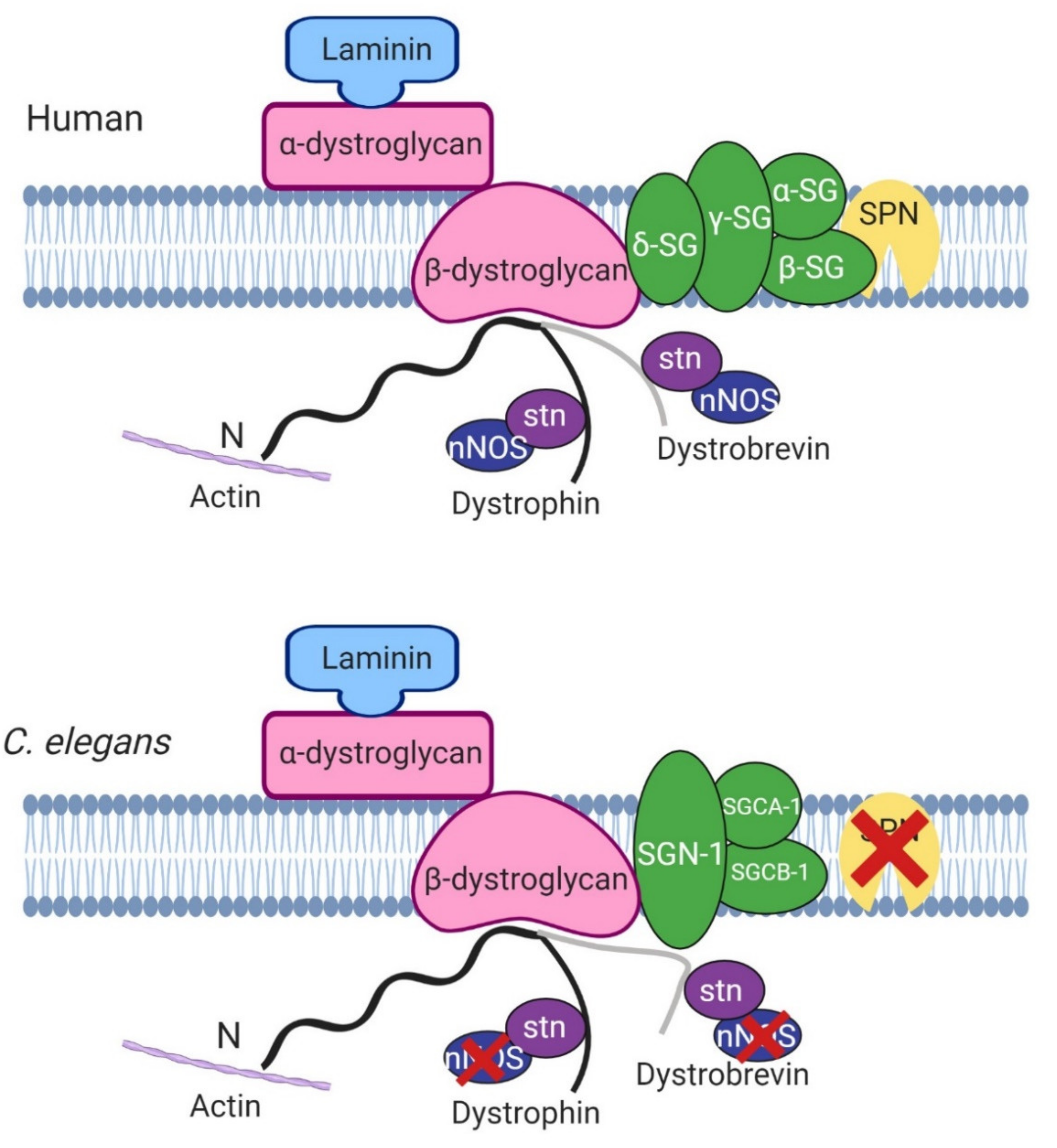
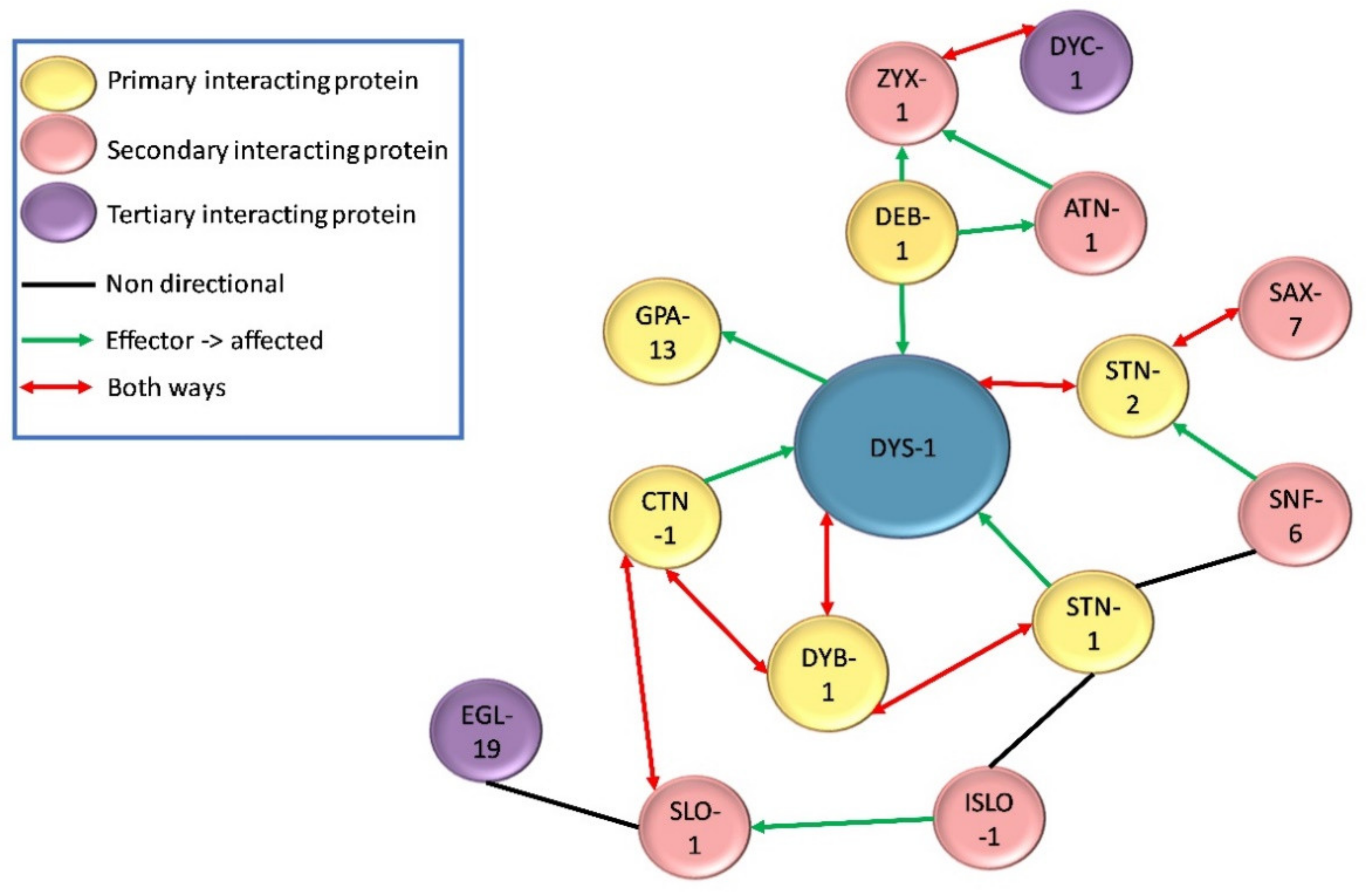
4. Physical Protein Interactions with DYS-1
4.1. DGC Associated Proteins
4.2. Dense Body Signalling Proteins
4.3. Calcium Homeostasis Associated Proteins
5. Genetic Interactions with dys-1
5.1. Dystrophin-Like Genes
5.2. Muscle Related Genes
5.3. Calcium Related Genes
5.4. Excitation–Contraction Coupling Genes
5.5. Mitochondrial Genes
5.6. Other Signalling Genes
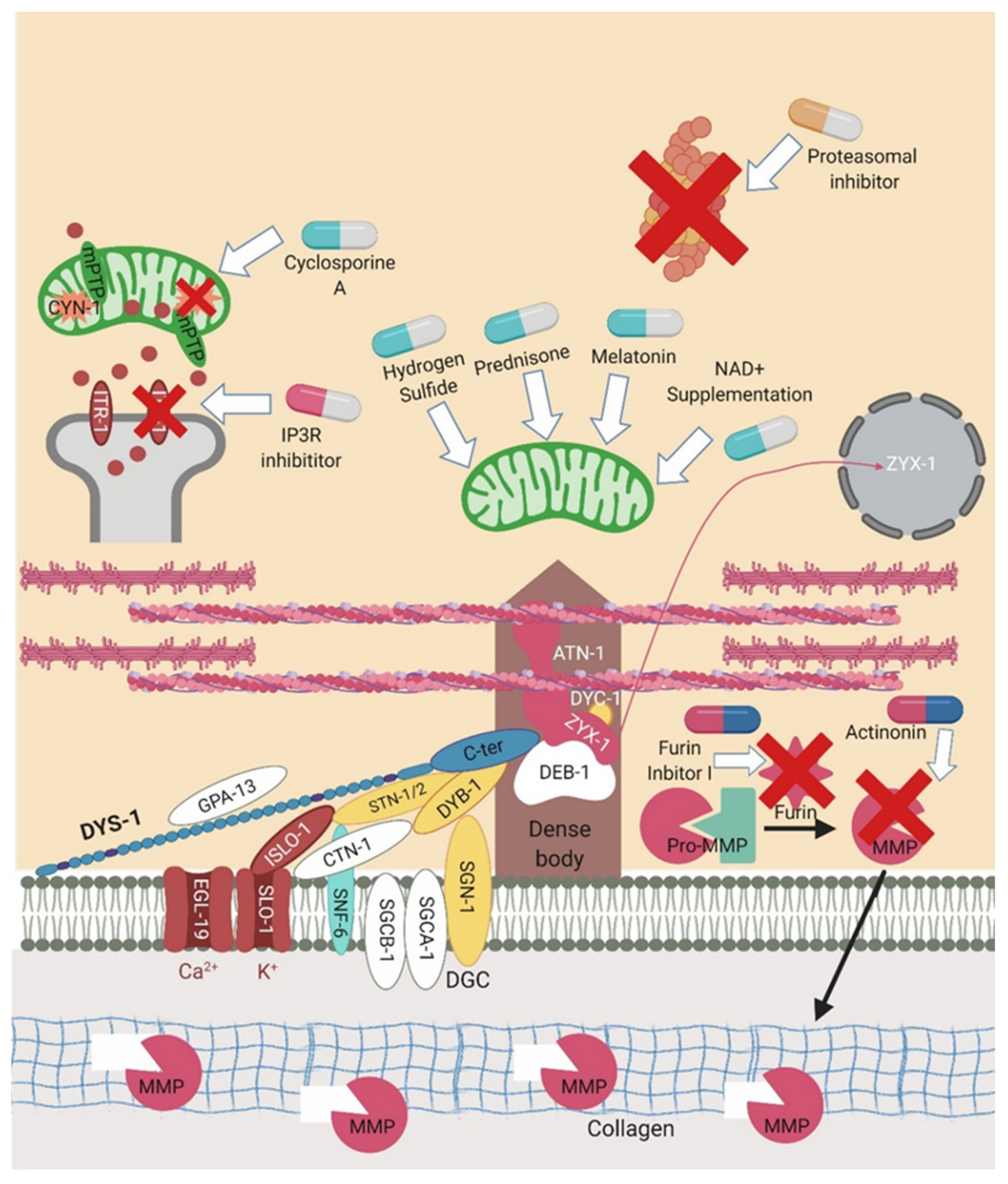
6. Pharmacological Interventions Trialled in dys-1 Mutants

6.1. Glucocorticoids
6.2. Hormone Related Therapies
6.3. Proteasome Inhibitors
6.4. Sulphonamides
6.5. Compounds Targeting the Mitochondria
6.6. Extracellular Matrix Targeted Compounds
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rumeur, E.L. Dystrophin and the two related genetic diseases, duchenne and becker muscular dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, J.; Gordon, K.E.; Dodds, L.; MacSween, J. Duchenne muscular dystrophy: A 30-year population-based incidence study. Clin. Pediatr. 2010, 49, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2015, 82, 291–329. [Google Scholar] [CrossRef] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Larcher, T.; Lafoux, A.; Tesson, L.; Remy, S.V.; Thepenier, V.; François, V.; Guiner, C.L.; Goubin, H.; Dutilleul, M.V.; Guigand, L.; et al. Characterization of dystrophin deficient rats: A new model for duchenne muscular dystrophy. PLoS ONE 2014, 9, e110371. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci. Rep. 2015, 4, 1–6. [Google Scholar] [CrossRef]
- Nguyen, F.; Cherel, Y.; Guigand, L.; Goubault-Leroux, I.; Wyers, M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J. Comp. Pathol. 2002, 126, 100–108. [Google Scholar] [CrossRef]
- Giugia, J.B.; Gieseler, K.; Arpagaus, M.; Ségalat, L. Mutations in the dystrophin-like Dys-1 gene of Caenorhabditis elegans result in reduced acetylcholinesterase activity. FEBS Lett. 1999, 463, 270–272. [Google Scholar] [CrossRef] [Green Version]
- Sleigh, J.N.; Sattelle, D.B.C. Elegans models of neuromuscular diseases expedite translational research. Transl. Neurosci. 2010, 1, 214–227. [Google Scholar] [CrossRef]
- Grisoni, K.; Martin, E.; Gieseler, K.; Mariol, M.C.; Ségalat, L. Genetic evidence for a Dystrophin-Glycoprotein Complex (DGC) in Caenorhabditis elegans. Gene 2002, 294, 77–86. [Google Scholar] [CrossRef]
- Hewitt, J.E.; Pollard, A.K.; Lesanpezeshki, L.; Deane, C.S.; Gaffney, C.J.; Etheridge, T.; Szewczyk, N.J.; Vanapalli, S.A. Muscle strength deficiency and mitochondrial dysfunction in a muscular dystrophy model of Caenorhabditis elegans and its functional response to drugs. Dis. Model. Mech. 2018, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ellwood, R.A.; Hewitt, J.E.; Torregrossa, R.; Philp, A.M.; Hardee, J.P.; Hughes, S.; van de Klashorse, D.; Gharahdaghi, N.; Anupom, T.; Slade, L.; et al. Mitochondrial hydrogen sulfide supplementation improves health in the C. elegans Duchenne muscular dystrophy model. Proc. Natl. Acad. Sci. USA 2021, 118, e2018342118. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.H.; Kim, H. Reduced IGF Signaling Prevents Muscle Cell Death in a Caenorhabditis Elegans Model of Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 19024–19029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altun, Z.F.; Hall, D.H. Pericellular structures. In WormAtlas; The C. elegans Research Community: Pasadena, CA, USA, 2009. [Google Scholar]
- Gieseler, K.; Qadota, H.; Benian, G.M. Development, structure, and maintenance of C. elegans body wall muscle. In WormBook; The C. elegans Research Community: Pasadena, CA, USA, 2017; pp. 1–59. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.P.; Dodd, A.; Overall, R.; Sirey, T.; Lam, L.T.; Morris, G.E.; Love, D.R. Dystrophin in adult zebrafish muscle. Biochem. Biophys. Res. Commun. 2001, 286, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Mosley, A.N.; Zhou, Y.; O’Brien, K.F.; Sheng, X.; Chiang, K.; Davidson, A.J.; Volinski, J.M.; Zon, L.I.; Kunkel, M. The dystrophin associated protein complex in zebrafish. Hum. Mol. Genet. 2003, 12, 601–615. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Chiyo, T.; Nishiyama, A.; Okada, H.; Takeda, S.; Okada, T. Dystrophic Mdx mice develop severe cardiac and respiratory dysfunction following genetic ablation of the anti-inflammatory cytokine IL-10. Hum. Mol. Genet. 2014, 23, 3990–4000. [Google Scholar] [CrossRef] [Green Version]
- Mcgreevy, J.W.; Hakim, C.H.; Mcintosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Models Mech. 2015, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Bessou, C.; Giugia, J.B.; Franks, C.J.; Holden-Dye, L.; Ségalat, L. Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics 1998, 2, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Krajacic, P.; Shen, X.; Purohit, P.K.; Arratia, P.; Lamitina, T. Biomechanical profiling of Caenorhabditis elegans motility. Genetics 2012, 191, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, C.; Schuler, A.; Vidal-Gadea, A.G. Method for the assessment of neuromuscular integrity and burrowing choice in vermiform animals. J. Neurosci. Methods 2016, 264, 40–46. [Google Scholar] [CrossRef]
- Beron, C.; Vidal-Gadea, A.G.; Cohn, J.; Parikh, A.; Huong, G.; Pierce-Shimomura, J.T. The burrowing behavior of the nematode Caenorhabditis elegans: A new assay for the study of neuromuscular disorders. Genes Brain Behav. 2015, 14, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Gieseler, K.; Grisoni, K.; Segalat, L. Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr. Biol. 2000, 10, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Sznitman, J.; Purohit, P.K.; Krajacic, P.; Lamitina, T.; Arratia, P.E. Material properties of Caenorhabditis elegans Swimming at low reynolds number. Biophys. J. 2010, 98, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, J.E.; Laranjeiro, R.; Norouzi, M.; Ellwood, R.A.; Antebi, A.; Szewczyk, N.J.; Driscoll, M.; Vanapalli, S.A. Multi-Environment phenotyping of C. elegans for robust evaluation of physical performance. bioRxiv 2020. [Google Scholar] [CrossRef]
- Scholtes, C.; Bellemin, S.; Martin, E.; Carre-Pierrat, M.; Mollereau, B.; Gieseler, K.; Walter, L. DRP-1-Mediated apoptosis induces muscle degeneration in dystrophin mutants. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Hrach, H.C.; O’Brien, S.; Steber, H.S.; Newbern, J.; Rawls, A.; Mangone, M. Transcriptome changes during the initiation and progression of duchenne muscular dystrophy in C. elegans. Hum. Mol. Genet. 2020, 29, 1607–1623. [Google Scholar] [CrossRef] [Green Version]
- Gieseler, K.; Abdel-Dayem, M.; Ségalat, L. In vitro interactions of Caenorhabditis elegans dystrophin with dystrobrevin and syntrophin. FEBS Lett. 1999, 461, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.S.; Benian, G.M. Muscular dystrophy: The worm turns to genetic disease. Curr. Biol. 2000, 10, 795–797. [Google Scholar] [CrossRef] [Green Version]
- Culetto, E.; Sattelle, D.B. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Mango, S.E. The C. elegans pharynx: A model for organogenesis. In Wormbook; The C. elegans Research Community: Pasadena, CA, USA, 2007. [Google Scholar] [CrossRef]
- Towers, P.R.; Lescure, P.; Baban, D.; Malek, J.A.; Duarte, J.; Jones, E.; Davies, K.E.; Ségalat, L.; Sattelle, D.B. Gene expression profiling studies on Caenorhabditis elegans dystrophin mutants Dys-1(Cx-35) and Dys-1(Cx18). Genomics 2006, 88, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in duchenne muscular dystrophy. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laumonier, T.; Menetrey, J. Muscle injuries and strategies for improving their repair. J. Exp. Orthop. 2016, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression Profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef]
- Gieseler, K.; Bessou, C.; Ségalat, L. Dystrobrevin- and Dystrophin-like mutants display similar phenotypes in the nematode Caenorhabditis elegans. Neurogenetics 1999, 2, 87–90. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, L. Neural integrity is maintained by dystrophin in C. elegans. J. Cell Biol. 2011, 192, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.P.C. Elegans dystroglycan DGN-1 functions in epithelia and neurons, but not muscle, and independently of dystrophin. Development 2006, 133, 1911–1921. [Google Scholar] [CrossRef]
- Blazie, S.M.; Babb, C.; Wilky, H.; Rawls, A.; Park, J.G.; Mangone, M. Comparative RNA-seq analysis reveals pervasive tissue-specific alternative polyadenylation in Caenorhabditis elegans intestine and muscles. BMC Biol. 2015, 13, 1–20. [Google Scholar] [CrossRef]
- Blazie, S.M.; Geissel, H.C.; Wilky, H.; Joshi, R.; Newbern, J.; Mangone, M. Alternative Polyadenylation directs tissue-specific MiRNA targeting in Caenorhabditis elegans somatic tissues. Genetics 2017, 206, 757–774. [Google Scholar] [CrossRef] [Green Version]
- Cuppen, E.; van der Linden, A.M.; Jansen, G.; Plasterk, R.H.A. Proteins Interacting with Caenorhabditis elegans Gα Subunits. Comp. Funct. Genom. 2003, 4, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.J.; Abraham, L.S.; Van Hengel, J.; Stove, C.; Proszynski, T.J.; Gevaert, K.; DiMario, J.X.; Sanes, J.R.; Van Roy, F.; Kim, H. Interaction of α-Catulin with dystrobrevin contributes to integrity of dystrophin complex in muscle. J. Biol. Chem. 2012, 287, 21717–21728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecroisey, C.; Brouilly, N.; Qadota, H.; Mariol, M.-C.; Rochette, N.C.; Martin, E.; Benian, G.M.; Segalat, L.; Mounier, N.; Gieseler, K. ZYX-1, the unique zyxin protein of Caenorhabditis elegans, is involved in dystrophin-dependent muscle degeneration. Mol. Biol. Cell 2013, 24, 1232–1249. [Google Scholar] [CrossRef] [PubMed]
- Brouilly, N.; Lecroisey, C.; Martin, E.; Pierson, L.; Mariol, M.C.; Mounier, N.; Gieseler, K.; Qadota, H.; Labouesse, M.; Streichenberger, N. Ultra-Structural time-course study in the C. elegans Model for duchenne muscular dystrophy highlights a crucial role for sarcomere-anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Hum. Mol. Genet. 2015, 24, 6428–6445. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Rogers, M.J.; Richmond, J.E.; McIntire, S.L. SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature 2004, 430, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Lecroisey, C.; Martin, E.; Mariol, M.C.; Granger, L.; Schwab, Y.; Labouesse, M.; Segalat, L.; Gieseler, K. DYC-1, a protein functionally linked to dystrophin in Caenorhabditis elegans is associated with the dense body, where it interacts with the muscle LIM domain protein ZYX-1. Mol. Biol. Cell 2008, 19, 785–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallejo-Illarrameni, A.; Toral-Ojeda, I.; Aldanondo, G.; Lopez de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [Green Version]
- Hughes, K.J.; Rodriguez, A.; Flatt, K.M.; Ray, S.; Schuler, A.; Rodemoyer, B.; Veerappan, V.; Cuciarone, K.; Kullman, A.; Lim, C.; et al. Physical exertion exacerbates decline in the musculature of an animal model of duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2019, 116, 3508–3517. [Google Scholar] [CrossRef] [Green Version]
- Carre-Pierrat, M.; Grisoni, K.; Gieseler, K.; Mariol, M.C.; Martin, E.; Jospin, M.; Allard, B.; Ségalat, L. The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J. Mol. Biol. 2006, 358, 387–395. [Google Scholar] [CrossRef]
- Kim, H.; Pierce-Shimomura, J.T.; Oh, H.J.; Johnson, B.E.; Goodman, M.B.; McIntire, S.L. The dystrophin complex controls BK channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000780. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Liu, P.; Wang, S.J.; Ge, Q.; Zhan, H.; Mohler, W.A.; Wang, Z.-W. A-Catulin CTN-1 is required for BK channel subcellular localization in C. Elegans body-wall muscle cells. EMBO 2010, 29, 3184–3195. [Google Scholar] [CrossRef] [Green Version]
- Gieseler, K.; Mariol, M.C.; Bessou, C.; Migaud, M.; Franks, C.J.; Holden-Dye, L.; Segalat, L. Molecular, genetic and physiological characterisation of dystrobrevin-like (Dyb-1) mutants of Caenorhabditis elegans. J. Mol. Biol. 2001, 307, 107–117. [Google Scholar] [CrossRef]
- Grisoni, K.; Gieseler, K.; Mariol, M.C.; Martin, E.; Carre-Pierrat, M.; Moulder, G.; Barstead, R.; Ségalat, L. The Stn-1 Syntrophin gene of C. elegans is functionally related to dystrophin and dystrobrevin. J. Mol. Biol. 2003, 332, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Jattujan, P.; Meemon, K.; Suphamungmee, W. Loss of dystrobrevin causes muscle degeneration and a short lifespan in Caenorhabditis elegans. Walailak J. Sci. Technol. 2018, 15, 659–667. [Google Scholar]
- Gieseler, K.; Grisoni, K.; Mariol, M.C.; Ségalat, L. Overexpression of dystrobrevin delays locomotion defects and muscle degeneration in a dystrophin-deficient Caenorhabditis elegans. Neuromuscul. Disord. 2002, 12, 371–377. [Google Scholar] [CrossRef]
- Mariol, M.C.; Martin, E.; Chambonnier, L.; Ségalat, L. Dystrophin-Dependent muscle degeneration requires a fully functional contractile machinery to occur in C. elegans. Neuromuscul. Disord. 2007, 17, 56–60. [Google Scholar] [CrossRef]
- Karpati, G.; Carpenter, S.; Prescott, S. Small-Caliber skeletal muscle fibers do not suffer necrosis in mdx mouse dystrophy. Muscle Nerve 1988, 11, 795–803. [Google Scholar] [CrossRef]
- Mizuno, Y. Prevention of myonecrosis in mdx mice: Effect of immobilization by the local tetanus method. Brain Dev. 1992, 14, 319–322. [Google Scholar] [CrossRef]
- Mokhtarian, A.; Lefaucheur, J.P.; Even, P.C.; Sebille, A. Hindlimb immobilization applied to 21-day-old mdx mice prevents the occurrence of muscle degeneration. J. Appl. Physiol. 1999, 86, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Bodeinsteiner, J.B.; Engel, A.G. Intracellular calcium accumulation in duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology 1978, 28, 439–446. [Google Scholar] [CrossRef]
- Zhan, H.; Stanciauskas, R.; Stigloher, C.; Dizon, K.K.; Jospin, M.; Bessereau, J.; Pinaud, F. In vivo single-molecule imaging identifies altered dynamics of calcium channels in dystrophin-mutant C. elegans. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mariol, M.C.; Ségalat, L. Muscular degeneration in the absence of dystrophin is a calcium-dependent process. Curr. Biol. 2001, 11, 1691–1694. [Google Scholar] [CrossRef] [Green Version]
- Sudevan, S.; Takiura, M.; Kubota, Y.; Higashitani, N.; Cooke, M.; Ellwood, R.A.; Etheridge, T.; Szewczyk, N.J.; Higashitani, A. Mitochondrial dysfunction causes Ca2+ overload and ECM degradation—Mediated muscle damage in C. elegans. FASEB J. 2019, 33, 9540–9550. [Google Scholar] [CrossRef] [PubMed]
- Giacomotto, J.; Brouilly, N.; Walter, L.; Mariol, M.C.; Berger, J.; Ségalat, L.; Becker, T.S.; Currie, P.D.; Gieseler, K. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and Ip3r activity in muscular dystrophy. Hum. Mol. Genet. 2013, 22, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Takagi, A.; Kojima, S.; Ida, M.; Araki, M. Increased leakage of calcium ion from the sarcoplasmic reticulum of the mdx mouse. J. Neurol. Sci. 1992, 110, 160–164. [Google Scholar] [CrossRef]
- Niebroj-Dobosz, I.; Kornguth, S.; Schutta, H.S.; Siegel, F.L. Elevated calmodulin levels and reduced calmodulin-stimulated calcium-ATPase in duchenne progressive muscular dystrophy. Neurology 1989, 39, 1610–1614. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Michel, S.A.; Chin, E.R.; Michel, R.N.; Jasmin, B.J. Targeted Inhibition of Ca2+/Calmodulin signaling exacerbates the dystrophic phenotype in mdx mouse muscle. Hum. Mol. Genet. 2006, 15, 1423–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briguet, A.; Erb, M.; Courdier-Fruh, I.; Barzaghi, P.; Santos, G.; Herzner, H.; Lescop, C.; Siendt, H.; Henneboehle, M.; Weyermann, P.; et al. Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 2008, 22, 4190–4200. [Google Scholar] [CrossRef]
- Joyce, P.I.; Satija, R.; Chen, M.; Kuwabara, P.E. The atypical calpains: Evolutionary analyses and roles in Caenorhabditis elegans cellular degeneration. PLoS Genet. 2012, 8, e1002602. [Google Scholar] [CrossRef] [Green Version]
- Ségalat, L.; Anderson, J.E. Duchenne muscular dystrophy: Stalled at the junction? Eur. J. Hum. Genet. 2005, 13, 4–5. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtsev, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Panel, M.; Lacôte, M.; Rieusset, J.; Lacampagne, A.; Fauconnier, J. Metformin reverses the enhanced myocardial SR/ER–mitochondria interaction and impaired complex I-driven respiration in dystrophin-deficient mice. Front. Cell Dev. Biol. 2021, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Lehner, B.; Crombie, C.; Wong, W.; Fraser, A.G.; Marcotte, E.M. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat. Genet. 2008, 40, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Nyamsuren, O.; Faggionato, D.; Loch, W.; Schulze, E.; Baumeister, R. A mutation in CHN-1/CHIP Suppresses muscle degeneration in Caenorhabditis elegans. Dev. Biol. 2007, 312, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomotto, J.; Pertl, C.; Borrel, C.; Walter, M.C.; Bulst, S.; Johnsen, B.; Baillie, D.L.; Lochmüller, H.; Thirion, C.; Ségalat, L. Evaluation of the therapeutic potential of carbonic anhydrase inhibitors in two animal models of dystrophin deficient muscular dystrophy. Hum. Mol. Genet. 2009, 18, 4089–4101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.Y.; Perreault, R.; Harel, S.; Boulier, E.L.; Suderman, M.; Hallett, M.; Jenna, S. Searching for signaling balance through the identification of genetic interactors of the rab guanine-nucleotide dissociation inhibitor Gdi-1. PLoS ONE 2010, 5, e10624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, L.P.; Luke, C.J.; Perlmutter, D.H.; Silverman, G.A.; Pak, S.C.C. Elegans in high-throughput drug discovery. Adv. Drug Deliv. Rev. 2013, 69–70, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Gaud, A.; Simon, J.M.; Witzel, T.; Carre-Pierrat, M.; Wermuth, C.G.; Ségalat, L. Prednisone reduces muscle degeneration in dystrophin-deficient Caenorhabditis elegans. Neuromuscul. Disord. 2004, 14, 365–370. [Google Scholar] [CrossRef]
- Carre-Pierrat, M.; Mariol, M.C.; Chambonnier, L.; Laugraud, A.; Heskia, F.; Giacomotto, J.; Ségalat, L. Blocking of striated muscle degeneration by serotonin in C. elegans. J. Muscle Res. Cell Motil. 2006, 27, 253–258. [Google Scholar] [CrossRef]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mazala, D.A.G.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in duchenne muscular dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Manning, J.; Kulbida, R.; Rai, P.; Jensen, L.; Bouma, J.; Singh, S.P.; O’Malley, D.; Yilmazer-Hanke, D.M. Amitriptyline is efficacious in ameliorating muscle inflammation and depressive symptoms in the mdx mouse model of duchenne muscular dystrophy. Exp. Physiol. 2014, 99, 1370–1386. [Google Scholar] [CrossRef] [PubMed]
- Waugh, T.A.; Horstick, E.; Hur, J.; Jackson, S.W.; Davidson, A.E.; Li, X.; Dowling, J.J. Fluoxetine prevents dystrophic changes in a zebrafish model of duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 4651–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, R.C.; Kuncl, R.W.; Morgan, J.; Cohen, L.; Meltzer, H.Y. Serotonin uptake in blood platelets of duchenne muscular dystrophy patients. Muscle Nerve 1987, 10, 359–362. [Google Scholar] [CrossRef]
- Murphy, D.L.; Mendell, J.R.; Engel, W.K. Serotonin and platelet function in duchenne muscular dystrophy. Arch. Neurol. 1973, 28, 239–242. [Google Scholar] [CrossRef]
- Chahbouni, M.; Escames, G.; Venegas, C.; Sevilla, B.; García, J.A.; López, L.C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuna-Castroviejo, D. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from duchenne muscular dystrophy. J. Pineal Res. 2010, 48, 282–289. [Google Scholar] [CrossRef]
- Chahbouni, M.; Escames, G.; López, L.C.; Sevilla, B.; Doerrier, C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment counteracts the hyperoxidative status in erythrocytes of patients suffering from duchenne muscular dystrophy. Clin. Biochem. 2011, 44, 853–858. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Reutenauer-Patte, J.; Patthey-Vuadens, O.; Ruegg, U.T.; Dorchies, O.M. Melatonin improves muscle function of the dystrophic mdx5Cv mouse, a model for duchenne muscular dystrophy. J. Pineal Res. 2011, 51, 163–171. [Google Scholar] [CrossRef]
- Talsness, D.M.; Belanto, J.J.; Ervasti, J.M. Disease-Proportional proteasomal degradation of missense dystrophins. Proc. Natl. Acad. Sci. USA 2015, 112, 12414–12419. [Google Scholar] [CrossRef] [Green Version]
- Assereto, S.; Stringara, S.; Sotiga, F.; Bonuccelli, G.; Broccolini, A.; Pedemonte, M.; Traverso, M.; Biancheri, R.; Zara, F.; Bruno, C.; et al. Pharmacological rescue of the dystrophin-glycoprotein complex in duchenne and becker skeletal muscle explants by proteasome inhibitor treatment. Am. J. Physiol. Cell Physiol. 2006, 290, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, T.; Fujimoto, S.; Horinouchi, H.; Ueyama, H.; Tsuda, T. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000, 100, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Sotgia, F.; Schubert, W.; Park, D.S.; Frank, P.G.; Woodman, S.E.; Insabato, L.; Cammer, M.; Minetti, C.; Lisanti, M.P. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am. J. Pathol. 2003, 163, 1663–1675. [Google Scholar] [CrossRef] [Green Version]
- Hollinger, K.; Selsby, J.T. The physiological response of protease inhibition in dystrophic muscle. Acta Physiol. 2013, 208, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.D.; Heath, R.; Jeffery, S.; Jackson, M.J.; Newham, D.J.; Edwards, R.H. Carbonic anhydrase III in duchenne muscular dystrophy. Clin. Chim. Acta 1983, 133, 201–208. [Google Scholar] [CrossRef]
- Ohta, M.; Itagaki, Y.; Itoh, N.; Hayashi, K.; Nishitani, H.; Ohta, K. Carbonic anhydrase III in serum in muscular dystrophy and other neurological disorders: Relationship with creatine kinase. Clin. Chem. 1991, 37, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Hor, K.N.; Mazur, W.; He, X.; Kissel, J.T.; Smart, S.; McCarthy, B.; Roble, S.L.; Cripe, L.H. Eplerenone for early cardiomyopathy in duchenne muscular dystrophy: Results of a two-year open-label extension trial. Orphanet J. Rare Dis. 2017, 12, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomotto, J.; Ségalat, L.; Carre-Pierrat, M.; Gieseler, K. Caenorhabditis elegans as a chemical screening tool for the study of neuromuscular disorders. Manual and semi-automated methods. Methods 2012, 56, 103–113. [Google Scholar] [CrossRef]
- De Luca, A.; Nico, B.; Liantonio, A.; Didonna, M.P.; Fraysse, B.; Pierno, S.; Burdi, R.; Mangieri, D.; Rolland, J.F.; Camerino, C.; et al. A Multidisciplinary evaluation of the effectiveness of cyclosporine A in dystrophic mdx mice. Am. J. Pathol. 2005, 166, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, J.; Schessl, J.; Schara, U.; Reitter, B.; Stettner, G.M.; Hobbiebrunken, E.; Wilichowski, E.; Bernert, G.; Weiss, S.; Stehling, F.; et al. Treatment of duchenne muscular dystrophy with ciclosporin A: A randomised, double-blind, placebo-controlled multicentre trial. Lancet Neurol. 2010, 9, 1053–1059. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 Receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Type | Benefits | Similarities to DMD in Humans | Limitations |
|---|---|---|---|
| C. elegans | Easy and cheap to maintain, short lifespan, high throughput experiments possible. Similar muscle structure and has orthologues for most human DGC proteins [10]. | Display movement and strength decline [11], altered gait [12] and shortened lifespan [13]. | Have a very simple body plan and nonconventional circulatory system [14]. Are unable to regenerate muscle as they lack satellite cells and do not have a conventional inflammatory system [15]. |
| Zebrafish | Easy to house and care for, high throughput experiments possible. High skeletal muscle content and expresses orthologues of most human DGC proteins [16]. | Changes in gait and lower activity [17]. | Missing several mammalian organs, are ectothermic and are influenced heavily by their environment. |
| Mdx mouse | One of the easier mammalian models to house and care for with a relatively short lifespan. High genetic similarity to humans including a DGC [4]. | Genetic and biochemical homologue of disease in humans. Displays ECG abnormalities and cardiomyopathy [18]. | Minimal clinical symptoms (no loss of ambulation and muscle weakness is not displayed until ~15 months) and lifespan is not majorly reduced [19]. |
| Dystrophin deficient rats | A convenient size as they are larger than mice allowing for studies with high statistical power but still relatively easy to house and care for. High genetic similarity including a DGC [5]. | Muscles showed severe fibrosis, muscle weakness and reduced activity [5,6]. | Not a well-established model and characterisation is still ongoing. |
| Golden retriever | Higher genetic similarity to humans compared to other mammalian models. Case reports showing that DMD occurs naturally in these animals as well. | Extensive homology in pathogenesis. Pathogenesis manifests in utero and extensive muscle necrosis can be seen and is progressive. They also have a shortened life span frequently dying from cardiac and respiratory failure [7]. | Expensive to maintain, not easily genetically manipulable and many ethical concerns. |
| Class of Phenotype | dys-1(cx18) | dys-1(cx18;hlh-1) | dys-1(eg33) |
|---|---|---|---|
| Locomotion | Exaggerated body bends, hyperactive, hypercontracted, overbent, swimming defective and burrowing defective [11,20,21,22,23]. | Exaggerated body bends, hyperactive, hypercontracted, overbent and swimming defective [24,25]. | Exaggerated body bends, hyperactive, hypercontracted, overbent, swimming defective and burrowing defective [11,12,22,23,26]. |
| Muscle structure | Very little muscle degeneration [20]. | Severe muscle degeneration [24]. | Severe muscle degeneration [13]. |
| Response to neuromuscular agents | Aldicarb hypersensitive, levamisole resistant [11,20]. | ND | Levamisole resistant [11]. |
| Mitochondria structure and function | Minor fragmentation of the mitochondria network, moderate depolarisation of mitochondrial membrane, no change in basal oxygen consumption rate [11]. | Severe fragmentation of the mitochondria network [27]. | Severe fragmentation of the mitochondria network, severe depolarisation of the mitochondrial membrane, elevated basal oxygen consumption rate [11,12]. |
| Life span | Shortened life span [13,28]. | ND | Shortened life span [12,13,28]. |
| Egg laying | No defect noted [20]. | Egg laying defect [24]. | ND |
| Strength | Not detectably weaker than WT [11]. | ND | Significant weakness detected compared to WT [11,12]. |
| Gene Classification | Associated Genes |
|---|---|
| Dystrophin-Like | dyb-1, dyc-1, islo-1, snf-6, slo-1, sgn-1, stn-1/2 |
| Muscle related | atn-1, lev-11, pat-10, unc-22, unc-89, unc-96, zyx-1 |
| Calcium | clp-1, cmd-1, csq-1, egl-19, islo-1, itr-1, sca-1, slo-1, stn-1, unc-2, unc-36, unc-68 |
| Excitation–contraction coupling | ace-1, ace-2, snf-6, unc-13, unc-29, unc-38 |
| Mitochondria | ced-1, ced-3, cps-6, crn-2, cyc-2.1, cyn-1, drp-1, eat-3, fzo-1, itr-1, psr-1, wah-1 |
| Other signalling | daf-2, daf-16, gst-4, let-60 |
| Other | cah-4; chn-1, gdi-1, hlh-1 |
| Drug Class | Tested Models | Proposed Mechanism of Action |
|---|---|---|
| Glucocorticoids (Prednisone) |  | Unknown hypothesised to have a direct effect on striated muscles (likely by repairing dysfunctional mitochondria and the mitochondrial network) [11,26,82]. |
| Serotonin |  | Unknown- as lack of dys-1 is known to disrupt signalling pathways it could affect serotonin receptors and by replacing the serotonin you can reduce muscle degeneration [11,26,83]. |
| Proteasomal inhibitor (MG132) |  | Inhibition of the proteasome rescues the protein localisation of the members of the DGC [78]. |
| Sulphonamides (methazolamide and dichlorphenamide) | | Inhibits cah-4 [79]. |
| Cyclosporine A |  | Inhibits cyn-1 which blocks or delays mPTP opening [66]. |
| IP3R inhibitor aminoethoxydiphenyl borate |  | Inhibits itr-1 [66]. |
| Nicotinamide riboside supplementation |  | Increases NAD+ levels [84]. |
| Melatonin |  | Reduces oxidative stress [11]. |
| Furin inhibitor I | | Inhibits Furin [65]. |
| Actinonin | | Inhibits matrix metalloproteinases [65]. |
| Hydrogen sulphide | | Improve mitochondrial dysfunction [12]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellwood, R.A.; Piasecki, M.; Szewczyk, N.J. Caenorhabditis elegans as a Model System for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2021, 22, 4891. https://doi.org/10.3390/ijms22094891
Ellwood RA, Piasecki M, Szewczyk NJ. Caenorhabditis elegans as a Model System for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2021; 22(9):4891. https://doi.org/10.3390/ijms22094891
Chicago/Turabian StyleEllwood, Rebecca A., Mathew Piasecki, and Nathaniel J. Szewczyk. 2021. "Caenorhabditis elegans as a Model System for Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 22, no. 9: 4891. https://doi.org/10.3390/ijms22094891
APA StyleEllwood, R. A., Piasecki, M., & Szewczyk, N. J. (2021). Caenorhabditis elegans as a Model System for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 22(9), 4891. https://doi.org/10.3390/ijms22094891