
Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer


{kind=link}
Abstract
:1. Introduction
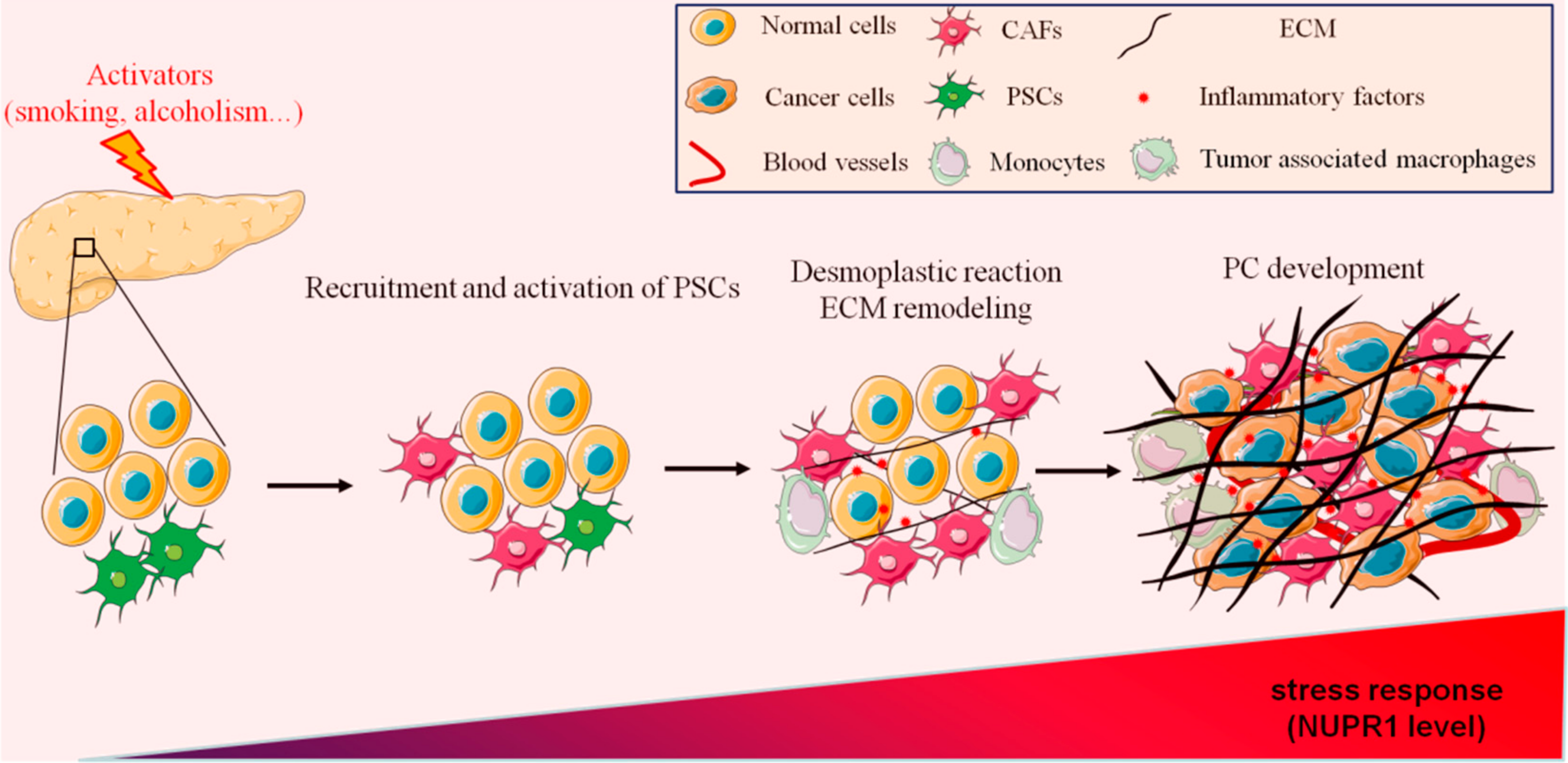
2. The Role of Fibrosis in Pancreatitis
2.1. Pancreatic Fibrosis Promotes Inflammation
2.2. Pancreatic Stellate Cells (PSCs) Are Key Mediators of Fibrosis in Pancreatitis
3. The Role of Fibrosis in PC
3.1. Pancreatic Fibrosis Promotes PC Progression
3.2. CAFs Contributes to Drug Resistance
3.2.1. Therapeutic Targeting of the Crosstalk between CAFs and PC
3.2.2. Targeting CAFs in Combination with Chemotherapy, a Field to Explore
3.2.3. CAFs Activation Suppresses Tumor Immune Response
3.2.4. CAFs Heterogeneity Is a Challenge in Cancer Therapy
4. Cellular Stress Response Led to Pancreatic Fibrosis
4.1. ROS Scavengers for Treating Pancreatic Fibrosis
4.2. Targeting Stress-Inducible Protein NUPR1 for Treating Pancreatic Fibrosis and PC
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| TP53 | Tumor protein p53 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| MRI | Magnetic resonance imaging |
| CT | Computerized tomography |
| CA199 | Carbohydrate antigen 199 |
| PC | Pancreatic cancer |
| CA125 | Cancer antigen 125 |
| GEM | Gemcitabine |
| CEA | Carcinoembryonic antigen |
| CA50 | Carbohydrate antigen 50 |
| CP | Chronic pancreatitis |
| AP | Acute pancreatitis |
| RECIST | response evaluation criteria in solid tumors |
| SPINK1 | Serine protease inhibitor Kazal-type 1 |
| PSCs | Pancreatic stellate cells |
| SST1 | symbiotic sulfate transporter 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| Hic-5 | Hydrogen peroxide-inducible clone-5 |
| PDAC | Pancreatic ductal adenocarcinoma |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| ACSL3 | Long-chain acyl coenzyme A synthase 3 |
| ADAM10 | A Disintegrin and metalloproteinase domain-containing protein 10 |
| CAFs | Cancer-related fibroblasts |
| FN | Fibronectin |
| SMO | Smoothened |
| FGFR | Fibroblast growth factor receptor |
| Hh | Hedgehog |
| ER stress | Endoplasmic reticulum stress |
| CAV1 | Caveolin-1 |
| ACTA2 | Actin alpha 2 |
| Tregs | Regulatory T cells |
| MDSCs | Myeloid-derived suppressor cells |
| myCAFs | Myofibroblastic CAFs |
| iCAFs | Inflammatory CAFs |
| apCAFs | Antigen-presenting CAFs |
| NK cells | Natural killer cells |
| CDH11 | Cadherin 11 |
| SMAD4 | SMAD family member 4 |
| TME | Tumor microenvironment |
| NDRG1 | N-myc downstream regulated gene-1 |
| CM | Camostat mesilate |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| MIR | MicroRNA |
| ECM | Extracellular matrix |
| NFs | Normal fibroblasts |
| MEFs | Mouse embryonic fibroblasts |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IL-1α | Interleukin-1 alpha |
| IL-1β | Interleukin 1 beta |
| TCR | T cell antigen receptor |
| NET-G1 | Grade 1 neuroendocrine tumor |
| 4E-BP1 | Eukaryotic translation initiation factor 4E binding protein 1 |
| MHC II | Major histocompatibility complex II |
| ROS | Reactive oxidative species |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NUPR1 | Nuclear protein 1 |
| NAC | N-acetyl cysteine |
| SOD | Superoxide dismutase |
| CoQ10 | Coenzyme Q10 |
References
- Xiao, B.; Zhang, X.-M. Magnetic resonance imaging for acute pancreatitis. World J. Radiol. 2010, 2, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Waldo, S.W.; Rosario, H.S.; Penn, A.H.; Schmid-Schönbein, G.W. Pancreatic digestive enzymes are potent generators of mediators for leukocyte activation and mortality. Shock 2003, 20, 138–143. [Google Scholar] [CrossRef]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef] [Green Version]
- Grigorian, A.; Lin, M.Y.C.; de Virgilio, C. Severe Epigastric Pain with Nausea and Vomiting. Surgery 2019, 227–237. [Google Scholar]
- Restrepo, R.; Hagerott, H.E.; Kulkarni, S.; Yasrebi, M.; Lee, E.Y. Acute Pancreatitis in Pediatric Patients: Demographics, Etiology, and Diagnostic Imaging. Am. J. Roentgenol. 2016, 206, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, E.V.F.; Guerrero-Lozano, R. Acute pancreatitis and recurrent acute pancreatitis: An exploration of clinical and etiologic factors and outcomes. J. Pediatr. 2019, 95, 713–719. [Google Scholar] [CrossRef]
- Barry, K. Chronic Pancreatitis: Diagnosis and Treatment. Am. Fam. Physician 2018, 97, 385–393. [Google Scholar]
- Pham, A.; Forsmark, C. Chronic pancreatitis: Review and update of etiology, risk factors, and management. F1000Research 2018, 7, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, P.J.; Soós, A.; Tóth, E.; Ébert, A.; Venglovecz, V.; Márta, K.; Mátrai, P.; Mikó, A.; Bajor, J.; Sarlós, P.; et al. Evidence for diagnosis of early chronic pancreatitis after three episodes of acute pancreatitis: A cross-sectional multicentre international study with experimental animal model. Sci. Rep. 2021, 11, 1367. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Pandol, S.J.; Porcel, J.; Wilkens, L.R.; Le Marchand, L.; Pike, M.C.; Monroe, K.R. Prospective Study of Alcohol Drinking, Smoking, and Pancreatitis: The Multiethnic Cohort. Pancreas 2016, 45, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, U.A.; Issa, Y.; Hagenaars, J.C.; Bakker, O.J.; van Goor, H.; Nieuwenhuijs, V.B.; Bollen, T.L.; van Ramshorst, B.; Witteman, B.J.; Brink, M.A.; et al. Risk of Recurrent Pancreatitis and Progression to Chronic Pancreatitis After a First Episode of Acute Pancreatitis. Clin. Gastroenterol. Hepatol. 2016, 14, 738–746. [Google Scholar]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, F.; Kaissis, G.A.; Ziegelmayer, S.; Harder, F.; Schilling, C.; Yen, H.Y.; Steiger, K.; Weichert, W.; Schirren, R.; Demir, I.E.; et al. Prediction of Tumor Cellularity in Resectable PDAC from Preoperative Computed Tomography Imaging. Cancers 2021, 13. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Satoi, S.; Yamamoto, T.; Yamaki, S.; Sekimoto, M. The past, present, and future status of multimodality treatment for resectable/borderline resectable pancreatic ductal adenocarcinoma. Surg. Today 2020, 50, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Srisajjakul, S.; Prapaisilp, P.; Bangchokdee, S. CT and MR features that can help to differentiate between focal chronic pancreatitis and pancreatic cancer. Radiol. Med. 2020, 125, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Benini, L.; Cavallini, G.; Zordan, D.; Rizzotti, P.; Rigo, L.; Brocco, G.; Perobelli, L.; Zanchetta, M.; Pederzoli, P.; Scuro, L.A. A clinical evaluation of monoclonal (CA19-9, CA50, CA12-5) and polyclonal (CEA, TPA) antibody-defined antigens for the diagnosis of pancreatic cancer. Pancreas 1988, 3, 61–66. [Google Scholar] [CrossRef]
- Goulden, M.R. The pain of chronic pancreatitis: A persistent clinical challenge. Br. J. Pain 2013, 7, 8–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kichler, A.; Jang, S. Chronic Pancreatitis: Epidemiology, Diagnosis, and Management Updates. Drugs 2020, 80, 1155–1168. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.L.; Conwell, D.L.; Hart, P.A. Complications of Chronic Pancreatitis. Dig. Dis. Sci. 2017, 62, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, G.; Habtezion, A.; Werner, J.; Lerch, M.M.; Mayerle, J. Chronic pancreatitis. Lancet 2020, 396, 499–512. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.; Wilson, J. The Fibrosis of Chronic Pancreatitis: New Insights into the Role of Pancreatic Stellate Cells. Antioxid. Redox Signal. 2011, 15, 2711–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, D.; DiVincenzo, M.J.; Torok, M.; Choueiry, F.; Kumar, R.J.; Deems, A.; Miller, J.L.; Hinton, A.; Geraghty, C.; Maranon, J.A.; et al. Soy-tomato enriched diet reduces inflammation and disease severity in a pre-clinical model of chronic pancreatitis. Sci. Rep. 2020, 10, 21824. [Google Scholar] [CrossRef]
- Di Magno, M.J.; Di Magno, E.P. Chronic pancreatitis. Curr. Opin. Gastroenterol. 2012, 28, 523–531. [Google Scholar] [CrossRef]
- Kanikovskiy, O.E.; Pavlyk, I.V.; Oliinyk, I.V.; Mosondz, V.V. The key role of pancreatic fibrosis severity in the surgical treatment algorithm of patients with chronic pancreatitis. Wiad. Lek. 2020, 73, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Witt, H. Chronic pancreatitis and cystic fibrosis. Gut 2003, 52, ii31–ii41. [Google Scholar] [CrossRef]
- Chowdhury, P.; Gupta, P. Pathophysiology of alcoholic pancreatitis: An overview. World J. Gastroenterol. 2006, 12, 7421–7427. [Google Scholar] [CrossRef]
- Lee, S.Y.; Goh, B.K.P.; Chan, C.Y. Chapter 55—Etiology, pathogenesis, and diagnostic assessment of acute pancreatitis. In Blumgart’s Surgery of the Liver, Biliary Tract and Pancreas; Jarnagin, W.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 883–896. [Google Scholar]
- Clemens, D.L.; Wells, M.A.; Schneider, K.J.; Singh, S. Molecular mechanisms of alcohol associated pancreatitis. World J. Gastrointest. Pathophysiol. 2014, 5, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, B.S.; Vasioukhin, V. Chapter 1—Mechanisms of Prostate Cancer Initiation and Progression. In Advances in Cancer Research; Woude, G.F.V., Klein, G., Eds.; Academic Press: London, UK, 2010; pp. 1–50. [Google Scholar]
- Jones, T.E.; Bellin, M.D.; Yadav, D.; Freeman, M.L.; Schwarzenberg, S.J.; Slivka, A.; Chennat, J.S.; Beilman, G.J.; Chinnakotla, S.; Pruett, T.L.; et al. The histopathology of SPINK1-associated chronic pancreatitis. Pancreatology 2020, 20, 1648–1655. [Google Scholar] [CrossRef]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E.; et al. Combined Bicarbonate Conductance-Impairing Variants in CFTR and SPINK1 Variants are Associated with Chronic Pancreatitis in Patients without Cystic Fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhakaran, P.R.; Radhika, A.; Jacob, S.S. Monocyte macrophage differentiation in vitro: Fibronectin-dependent upregulation of certain macrophage-specific activities. Glycoconj. J. 2007, 24, 49–55. [Google Scholar] [CrossRef]
- Mescher, A.L. Macrophages and fibroblasts during inflammation and tissue repair in models of organ regeneration. Regeneration 2017, 4, 39–53. [Google Scholar] [CrossRef]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of Immune Cells and Immune-Based Therapies in Pancreatitis and Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158-7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Lou, N.; Jiao, J.; Guo, F.; Xiang, H.; Shang, D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed. Pharmacother. 2020, 131, 110693. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Duan, L.; Wu, N.; Xu, X.; Xin, J.; Jiang, S.; Zhang, C.; Zhang, H. Baicalin Ameliorates Pancreatic Fibrosis by Inhibiting the Activation of Pancreatic Stellate Cells in Mice with Chronic Pancreatitis. Front. Pharmacol. 2020, 11, 607133. [Google Scholar] [CrossRef]
- Alpha, K.M.; Xu, W.; Turner, C.E. Chapter One—Paxillin family of focal adhesion adaptor proteins and regulation of cancer cell invasion. In International Review of Cell and Molecular Biology; Thomas, C., Galluzzi, L., Eds.; Academic Press: London, UK, 2020; pp. 1–52. [Google Scholar]
- Gao, L.; Lei, X.-F.; Miyauchi, A.; Noguchi, M.; Omoto, T.; Haraguchi, S.; Miyazaki, T.; Miyazaki, A.; Kim-Kaneyama, J.-R. Hic-5 is required for activation of pancreatic stellate cells and development of pancreatic fibrosis in chronic pancreatitis. Sci. Rep. 2020, 10, 19105. [Google Scholar] [CrossRef]
- Rasmussen, H.H.; Irtun, O.; Olesen, S.S.; Drewes, A.M.; Holst, M. Nutrition in chronic pancreatitis. World J. Gastroenterol. 2013, 19, 7267–7275. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Moneo, E.; Stigliano, S.; Hedstrom, A.; Kaczka, A.; Malvik, M.; Waldthaler, A.; Maisonneuve, P.; Simon, P.; Capurso, G. Deficiency of fat-soluble vitamins in chronic pancreatitis: A systematic review and meta-analysis. Pancreatology 2016, 16. [Google Scholar] [CrossRef]
- Du, W.D.; Yuan, Z.R.; Sun, J.; Tang, J.X.; Cheng, A.Q.; Shen, D.M.; Huang, C.J.; Song, X.H.; Yu, X.F.; Zheng, S.B. Therapeutic efficacy of high-dose vitamin C on acute pancreatitis and its potential mechanisms. World J. Gastroenterol. 2003, 9, 2565–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharer, N.M.; Taylor, P.M.; Linaker, B.D.; Gutteridge, J.M.C.; Braganza, J.M. Safe and Successful Use of Vitamin C to Treat Painful Calcific Chronic Pancreatitis Despite Iron Overload from Primary Haemochromatosis. Clin. Drug Investig. 1995, 10, 310–315. [Google Scholar] [CrossRef]
- Lu, X.L.; Song, Y.H.; Fu, Y.B.; Si, J.M.; Qian, K.D. Ascorbic Acid Alleviates Pancreatic Damage Induced by Dibutyltin Dichloride (DBTC) in Rats. Yonsei Med. J. 2007, 48, 1028–1034. [Google Scholar] [CrossRef]
- Al-Hashem, F.; Ellatif, M.A.; Shamseldeen, A.M.; Kamar, S.S.; Al-Ani, B.; Haidara, M.A. Vitamin E protects against the modulation of TNF-α-AMPK axis and inhibits pancreas injury in a rat model of L-arginine-induced acute necrotising pancreatitis. Arch. Physiol. Biochem. 2020, 1–9. [Google Scholar] [CrossRef]
- Reifen, R. Vitamin A as an anti-inflammatory agent. Proc. Nutr. Soc. 2002, 61, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Chen, H.; Qin, S.; Wang, M.; Wang, X.; Zhang, X.; Liu, F.; Zhang, S. The association between dietary vitamin A intake and pancreatic cancer risk: A meta-analysis of 11 studies. Biosci. Rep. 2016, 36, e00414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siener, R.; Machaka, I.; Alteheld, B.; Bitterlich, N.; Metzner, C. Effect of Fat-Soluble Vitamins A, D, E and K on Vitamin Status and Metabolic Profile in Patients with Fat Malabsorption with and without Urolithiasis. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Miyauchi, M.; Suda, K.; Kuwayama, C.; Abe, H.; Kakinuma, C. Role of fibrosis-related genes and pancreatic duct obstruction in rat pancreatitis models: Implications for chronic pancreatitis. Histol. Histopathol. 2007, 22, 1119–1127. [Google Scholar]
- Tseng, J.; Choi, E.A.; Matthews, J.B. Chapter 92—Chronic Pancreatitis. In Shackelford’s Surgery of the Alimentary Tract; Yeo, C.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1085–1096. [Google Scholar]
- Ramakrishnan, P.; Loh, W.M.; Gopinath, S.C.B.; Bonam, S.R.; Fareez, I.M.; Mac Guad, R.; Sim, M.S.; Wu, Y.S. Selective phytochemicals targeting pancreatic stellate cells as new anti-fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. Bad Neighborhood: Fibrotic Stroma as a New Player in Melanoma Resistance to Targeted Therapies. Cancers 2020, 12, 1364. [Google Scholar] [CrossRef] [PubMed]
- Peran, I.; Dakshanamurthy, S.; McCoy, M.D.; Mavropoulos, A.; Allo, B.; Sebastian, A.; Hum, N.R.; Sprague, S.C.; Martin, K.A.; Pishvaian, M.J.; et al. Cadherin 11 Promotes Immunosuppression and Extracellular Matrix Deposition to Support Growth of Pancreatic Tumors and Resistance to Gemcitabine in Mice. Gastroenterology 2021, 160. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Inoue, Y.; Hiratsuka, M.; Kawakatsu, S.; Arai, T.; Matsueda, K.; Saiura, A.; Takazawa, Y. Encapsulating fibrosis following neoadjuvant chemotherapy is correlated with outcomes in patients with pancreatic cancer. PLoS ONE 2019, 14, e0222155. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erstad, D.J.; Sojoodi, M.; Taylor, M.S.; Jordan, V.C.; Farrar, C.T.; Axtell, A.L.; Rotile, N.J.; Jones, C.; Graham-O’Regan, K.A.; Ferreira, D.S.; et al. Fibrotic Response to Neoadjuvant Therapy Predicts Survival in Pancreatic Cancer and Is Measurable with Collagen-Targeted Molecular MRI. Clin. Cancer Res. 2020, 26, 5007–5018. [Google Scholar] [CrossRef]
- Palomino, D.C.T.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Admyre, C.; Axelsson, L.-G.; von Stein, O.; Zargari, A. Immunomodulatory oligonucleotides inhibit neutrophil migration by decreasing the surface expression of interleukin-8 and leukotriene B4 receptors. Immunology 2015, 144, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Pozzato, C.; Saliakoura, M.; Yang, Z.; Peng, R.-W.; Galiè, M.; Oberson, K.; Simon, H.-U.; Karamitopoulou, E.; Konstantinidou, G. ACSL3-PAI-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Fàbrega, A.; Guyonnet, B.; Dacheux, J.L.; Gatti, J.L.; Puigmulé, M.; Bonet, S.; Pinart, E. Expression, immunolocalization and processing of fertilins ADAM-1 and ADAM-2 in the boar (Sus domesticus) spermatozoa during epididymal maturation. Reprod. Biol. Endocrinol. 2011, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.C.; Piper, M.; Goodspeed, A.; Bhuvane, S.; Williams, J.S.; Bhatia, S.; Phan, A.V.; Van Court, B.; Zolman, K.L.; Peña, B.; et al. Induction of ADAM10 by RT drives fibrosis, resistance, and EMT in pancreatic cancer. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; De Nardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Araki, K.; Yokobori, T.; Hagiwara, K.; Gantumur, D.; Yamanaka, T.; Handa, T.; Tsukagoshi, M.; Igarashi, T.; Watanabe, A.; et al. Conophylline suppresses pancreatic cancer desmoplasia and cancer-promoting cytokines produced by cancer-associated fibroblasts. Cancer Sci. 2019, 110, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife 2019, 8. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of Smoothened Activates the Sonic Hedgehog Signaling Pathway in Pancreatic Cancer-Associated Fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. NDRG1, a growth and cancer related gene: Regulation of gene expression and function in normal and disease states. Carcinogenesis 2008, 29, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geleta, B.; Park, K.C.; Jansson, P.J.; Sahni, S.; Maleki, S.; Xu, Z.; Murakami, T.; Pajic, M.; Apte, M.V.; Richardson, D.R.; et al. Breaking the cycle: Targeting of NDRG1 to inhibit bi-directional oncogenic cross-talk between pancreatic cancer and stroma. FASEB J. 2021, 35, e21347. [Google Scholar] [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Modica, C.; Tortarolo, D.; Comoglio, P.M.; Basilico, C.; Vigna, E. MET/HGF Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight into the Tumor/Stroma Crosstalk. Int. J. Mol. Sci. 2018, 19, 3920. [Google Scholar] [CrossRef] [Green Version]
- Basso, D.; Bozzato, D.; Padoan, A.; Moz, S.; Zambon, C.-F.; Fogar, P.; Greco, E.; Scorzeto, M.; Simonato, F.; Navaglia, F.; et al. Inflammation and pancreatic cancer: Molecular and functional interactions between S100A8, S100A9, NT-S100A8 and TGFβ1. Cell Commun. Signal. 2014, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers 2017, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Ning, Z.; Ma, L.; Liu, W.; Shao, C.; Shu, Y.; Shen, H. Exosomal miRNAs and miRNA dysregulation in cancer-associated fibroblasts. Mol. Cancer 2017, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Samain, R.; Brunel, A.; Douché, T.; Fanjul, M.; Cassant-Sourdy, S.; Rochotte, J.; Cros, J.; Neuzillet, C.; Raffenne, J.; Duluc, C.; et al. Pharmacological Normalization of Pancreatic Cancer-Associated Fibroblast Secretome Impairs Pro-metastatic Cross-Talk with Macrophages. Cell. Mol. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 2018, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Shi, S.; Meng, Q.; Liang, D.; Ji, S.; Zhang, B.; Qin, Y.; Xu, J.; Ni, Q.; Yu, X. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: Where we are and where we are going. Exp. Mol. Med. 2017, 49, e406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.-H. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Cui, J.Y.; Gao, H.F.; Yu, H.; Gao, F.F.; Chen, J.L.; Chen, L. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 2020, 16, 2619–2633. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-Associated Fibroblasts in Pancreatic Cancer: Should They Be Deleted or Reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef]
- Dauer, P.; Zhao, X.; Gupta, V.K.; Sharma, N.; Kesh, K.; Gnamlin, P.; Dudeja, V.; Vickers, S.M.; Banerjee, S.; Saluja, A. Inactivation of Cancer-Associated-Fibroblasts Disrupts Oncogenic Signaling in Pancreatic Cancer Cells and Promotes Its Regression. Cancer Res. 2018, 78, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Yamao, T.; Yamashita, Y.I.; Yamamura, K.; Nakao, Y.; Tsukamoto, M.; Nakagawa, S.; Okabe, H.; Hayashi, H.; Imai, K.; Baba, H. Cellular Senescence, Represented by Expression of Caveolin-1, in Cancer-Associated Fibroblasts Promotes Tumor Invasion in Pancreatic Cancer. Ann. Surg. Oncol. 2019, 26, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Vendramini-Costa, D.B.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast-Driven Nutritional Support and Immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Johnson, D.K.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013, 55, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.B.; Steele, N.G.; Carpenter, E.S.; Donahue, K.L.; Bushnell, G.G.; Morris, A.H.; The, S.; Orbach, S.M.; Sirihorachai, V.R.; Nwosu, Z.C.; et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance 2021, 4, e202000935. [Google Scholar] [CrossRef]
- Huang, H.; Brekken, R.A. Recent advances in understanding cancer-associated fibroblasts in pancreatic cancer. Am. J. Physiol. Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.-R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Colombo, M.; Mirandola, L.; Chiriva-Internati, M.; Basile, A.; Locati, M.; Lesma, E.; Chiaramonte, R.; Platonova, N. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Front. Immunol. 2018, 9, 1823-1823. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Zheng, L. Epigenetics in modulating immune functions of stromal and immune cells in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 940–953. [Google Scholar] [CrossRef]
- Freeman, P.; Mielgo, A. Cancer-Associated Fibroblast Mediated Inhibition of CD8+ Cytotoxic T Cell Accumulation in Tumours: Mechanisms and Therapeutic Opportunities. Cancers 2020, 12, 2687. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Brekken, R. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. bioRxiv 2021. bioRxiv:10.2139/ssrn.3790948. [Google Scholar]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Schuck, K.; Friess, H.; Kong, B. Targeting Aggressive Fibroblasts to Enhance the Treatment of Pancreatic Cancer. Expert Opin. Ther. Targets 2021, 25, 5–13. [Google Scholar] [CrossRef]
- Chovatiya, R.; Medzhitov, R. Stress, Inflammation, and Defense of Homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031-16031. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative Stress, Tumor Microenvironment, and Metabolic Reprogramming: A Diabolic Liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.-M.; Oh, T.-Y.; Kim, Y.-B.; Yeo, M.; Lee, J.-S.; Surh, Y.J.; Ahn, B.-O.; Kim, W.-H.; Sohn, S.; Kim, J.-H.; et al. Novel antioxidant ameliorates the fibrosis and inflammation of cerulein-induced chronic pancreatitis in a mouse model. Pancreatology 2005, 5, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Heras-Castaño, G.D.L.; García-Unzueta, M.T.; Domínguez-Diez, A.; Fernández-González, M.D.; la Paz, A.M.G.D.; Mayorga-Fernández, M.; Fernández-Fernández, F. Pancreatic fibrosis in rats and its response to antioxidant treatment. JOP 2005, 6, 316–324. [Google Scholar]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Yan, B.; Cheng, L.; Jiang, Z.; Chen, K.; Zhou, C.; Sun, L.; Cao, J.; Qian, W.; Li, J.; Shan, T.; et al. Resveratrol Inhibits ROS-Promoted Activation and Glycolysis of Pancreatic Stellate Cells via Suppression of miR-21. Oxidative Med. Cell. Longev. 2018, 2018, 1346958. [Google Scholar] [CrossRef]
- Gonzalez, A.; Estaras, M.; Martinez-Morcillo, S.; Martinez, R.; García, A.; Estévez, M.; Santofimia-Castaño, P.; Tapia, J.A.; Moreno, N.; Pérez-López, M.; et al. Melatonin modulates red-ox state and decreases viability of rat pancreatic stellate cells. Sci. Rep. 2020, 10, 6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estaras, M.; Moreno, N.; Santofimia-Castaño, P.; Martinez-Morcillo, S.; Roncero, V.; Blanco, G.; Lopez, D.; Fernandez-Bermejo, M.; Mateos, J.M.; Iovanna, J.L.; et al. Melatonin induces reactive oxygen species generation and changes in glutathione levels and reduces viability in human pancreatic stellate cells. J. Physiol. Biochem. 2019, 75, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kojayan, G.G.; Alizadeh, R.F.; Li, S.; Ichii, H. Reducing Pancreatic Fibrosis Using Antioxidant Therapy Targeting Nrf2 Antioxidant Pathway: A Possible Treatment for Chronic Pancreatitis. Pancreas 2019, 48, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Ghosh, S.; Gupta, P.; Mukherjee, S.; Chattopadhyay, S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic. Res. 2015, 49, 1371–1383. [Google Scholar] [CrossRef]
- Matsumura, N.; Ochi, K.; Ichimura, M.; Mizushima, T.; Harada, H.; Harada, M. Study on free radicals and pancreatic fibrosis--pancreatic fibrosis induced by repeated injections of superoxide dismutase inhibitor. Pancreas 2001, 22, 53–57. [Google Scholar] [CrossRef]
- Xu, M.; Cai, J.; Wei, H.; Zhou, M.; Xu, P.; Huang, H.; Peng, W.; Du, F.; Gong, A.; Zhang, Y. Scoparone Protects Against Pancreatic Fibrosis via TGF-β/Smad Signaling in Rats. Cell. Physiol. Biochem. 2016, 40, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Yang, J.; Wu, J.; Meng, Q.; Hao, J. Coenzyme Q10 inhibits the activation of pancreatic stellate cells through PI3K/AKT/mTOR signaling pathway. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Xue, R.; Wang, J.; Yang, L.; Liu, X.; Gao, Y.; Pang, Y.; Wang, Y.; Hao, J. Coenzyme Q10 Ameliorates Pancreatic Fibrosis via the ROS-Triggered mTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 8039694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallo, G.V.; Fiedler, F.; Calvo, E.L.; Ortiz, E.M.; Vasseur, S.; Keim, V.; Morisset, J.; Iovanna, J.L. Cloning and Expression of the Rat p8 cDNA, a New Gene Activated in Pancreas during the Acute Phase of Pancreatitis, Pancreatic Development, and Regeneration, and Which Promotes Cellular Growth. J. Biol. Chem. 1997, 272, 32360–32369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoo, Y.; Iovanna, J.L.; Mallo, G.V.; Su, S.B.; Xie, M.J.; Sawabu, N. P8 expression is induced in acinar cells during chronic pancreatitis. Dig. Dis. Sci. 2001, 46, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Motoo, Y.; Iovanna, J.L.; Berthézène, P.; Xie, M.J.; Mouri, H.; Ohtsubo, K.; Matsubara, F.; Sawabu, N. Overexpression of p8 is inversely correlated with apoptosis in pancreatic cancer. Clin. Cancer Res. 2001, 7, 1320–1324. [Google Scholar] [PubMed]
- Zhou, R.; Liao, J.; Cai, D.; Tian, Q.; Huang, E.; Lü, T.; Chen, S.Y.; Xie, W.B. Nupr1 mediates renal fibrosis via activating fibroblast and promoting epithelial-mesenchymal transition. FASEB J. 2021, 35, e21381. [Google Scholar] [CrossRef]
- Zhong, C.; Tao, B.; Tang, F.; Yang, X.; Peng, T.; You, J.; Xia, K.; Xia, X.; Chen, L.; Peng, L. Remodeling cancer stemness by collagen/fibronectin via the AKT and CDC42 signaling pathway crosstalk in glioma. Theranostics 2021, 11, 1991–2005. [Google Scholar] [CrossRef]
- Georgescu, S.P.; Aronovitz, M.J.; Iovanna, J.L.; Patten, R.D.; Kyriakis, J.M.; Goruppi, S. Decreased metalloprotease 9 induction, cardiac fibrosis, and higher autophagy after pressure overload in mice lacking the transcriptional regulator p8. Am. J. Physiol. Physiol. 2011, 301, C1046–C1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.-B.; Motoo, Y.; Iovanna, J.L.; Xie, M.-J.; Sawabu, N. Effect of Camostat Mesilate on the Expression of Pancreatitis-Associated Protein (PAP), p8, and Cytokines in Rat Spontaneous Chronic Pancreatitis. Pancreas 2001, 23, 134–140. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abián, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Xia, Y.; Zhou, Z.; Huang, C.; Fraunhoffer, N.; Barea, D.; Cervello, M.; Giannitrapani, L.; Montalto, G.; et al. Targeting NUPR1 with the small compound ZZW-115 is an efficient strategy to treat hepatocellular carcinoma. Cancer Lett. 2020, 486, 8–17. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Swayden, M.; Xia, Y.; Zhou, Z.; Audebert, S.; Camoin, L.; Huang, C.; Peng, L.; Jiménez-Alesanco, A.; et al. ZZW-115-dependent inhibition of NUPR1 nuclear translocation sensitizes cancer cells to genotoxic agents. JCI Insight 2020. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-knife of cancer. J. Cell. Physiol. 2011, 226, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Lan, W.; Bintz, J.; Gayet, O.; Carrier, A.; Lomberk, G.; Neira, J.L.; González, A.; Urrutia, R.; Soubeyran, P.; et al. Inactivation of NUPR1 promotes cell death by coupling ER-stress responses with necrosis. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, T.; Cano, C.E.; Grasso, D.; Garcia, M.N.; Sandi, M.J.; Calvo, E.L.; Dagorn, J.C.; Lomberk, G.; Goruppi, S.; Urrutia, R.; et al. NUPR1 works against the metabolic stress-induced autophagy-associated cell death in pancreatic cancer cells. Autophagy 2013, 9, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Santofimia-Castaño, P.; Iovanna, J. Combating pancreatic cancer chemoresistance by triggering multiple cell death pathways. Pancreatology 2021. [Google Scholar] [CrossRef]
- Adam, M.G.; Beyer, G.; Christiansen, N.; Kamlage, B.; Pilarsky, C.; Distler, M.; Falbusch, T.; Chromik, A.; Klein, F.; Bahra, M.; et al. Identification and validation of a multivariable prediction model based on blood plasma and serum metabolomics for the distinction of chronic pancreatitis subjects from non-pancreas disease control subjects. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Novovic, S.; Borch, A.; Werge, M.; Karran, D.; Gluud, L.; Schmidt, P.N.; Hansen, E.F.; Nøjgaard, C.; Jensen, A.B.; Jensen, F.K.; et al. Characterisation of the fibroinflammatory process involved in progression from acute to chronic pancreatitis: Study protocol for a multicentre, prospective cohort study. BMJ Open 2019, 9, e028999. [Google Scholar] [CrossRef] [Green Version]
- Oppong, K.W.; Bekkali, N.L.H.; Leeds, J.S.; Johnson, S.J.; Nayar, M.K.; Darné, A.; Egan, M.; Bassett, P.; Haugk, B. Fork-tip needle biopsy versus fine-needle aspiration in endoscopic ultrasound-guided sampling of solid pancreatic masses: A randomized crossover study. Endoscopy 2020, 52, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Crinò, S.F.; Le Grazie, M.; Manfrin, E.; Bellocchi, M.C.C.; Bernardoni, L.; Granato, A.; Locatelli, F.; Parisi, A.; Di Stefano, S.; Frulloni, L.; et al. Randomized trial comparing fork-tip and side-fenestrated needles for EUS-guided fine-needle biopsy of solid pancreatic lesions. Gastrointest. Endosc. 2020, 92, 648–658. [Google Scholar] [CrossRef]
- Kandel, P.; Nassar, A.; Gomez, V.; Raimondo, M.; Woodward, T.A.; Crook, J.E.; Fares, N.S.; Wallace, M.B. Comparison of endoscopic ultrasound-guided fine-needle biopsy versus fine-needle aspiration for genomic profiling and DNA yield in pancreatic cancer: A randomized crossover trial. Endoscopy 2021, 53, 376–382. [Google Scholar]
- Hollinshead, K.E.R.; Parker, S.J.; Eapen, V.V.; Encarnacion-Rosado, J.; Sohn, A.; Oncu, T.; Cammer, M.; Mancias, J.D.; Kimmelman, A.C. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep. 2020, 33, 108231. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Iovanna, J.; Santofimia-Castaño, P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 4970. https://doi.org/10.3390/ijms22094970
Huang C, Iovanna J, Santofimia-Castaño P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. International Journal of Molecular Sciences. 2021; 22(9):4970. https://doi.org/10.3390/ijms22094970
Chicago/Turabian StyleHuang, Can, Juan Iovanna, and Patricia Santofimia-Castaño. 2021. "Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer" International Journal of Molecular Sciences 22, no. 9: 4970. https://doi.org/10.3390/ijms22094970
APA StyleHuang, C., Iovanna, J., & Santofimia-Castaño, P. (2021). Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. International Journal of Molecular Sciences, 22(9), 4970. https://doi.org/10.3390/ijms22094970