
The Plasminogen–Activator Plasmin System in Physiological and Pathophysiological Angiogenesis


Abstract
:1. Introduction
2. Plasminogen Activator–Plasmin System
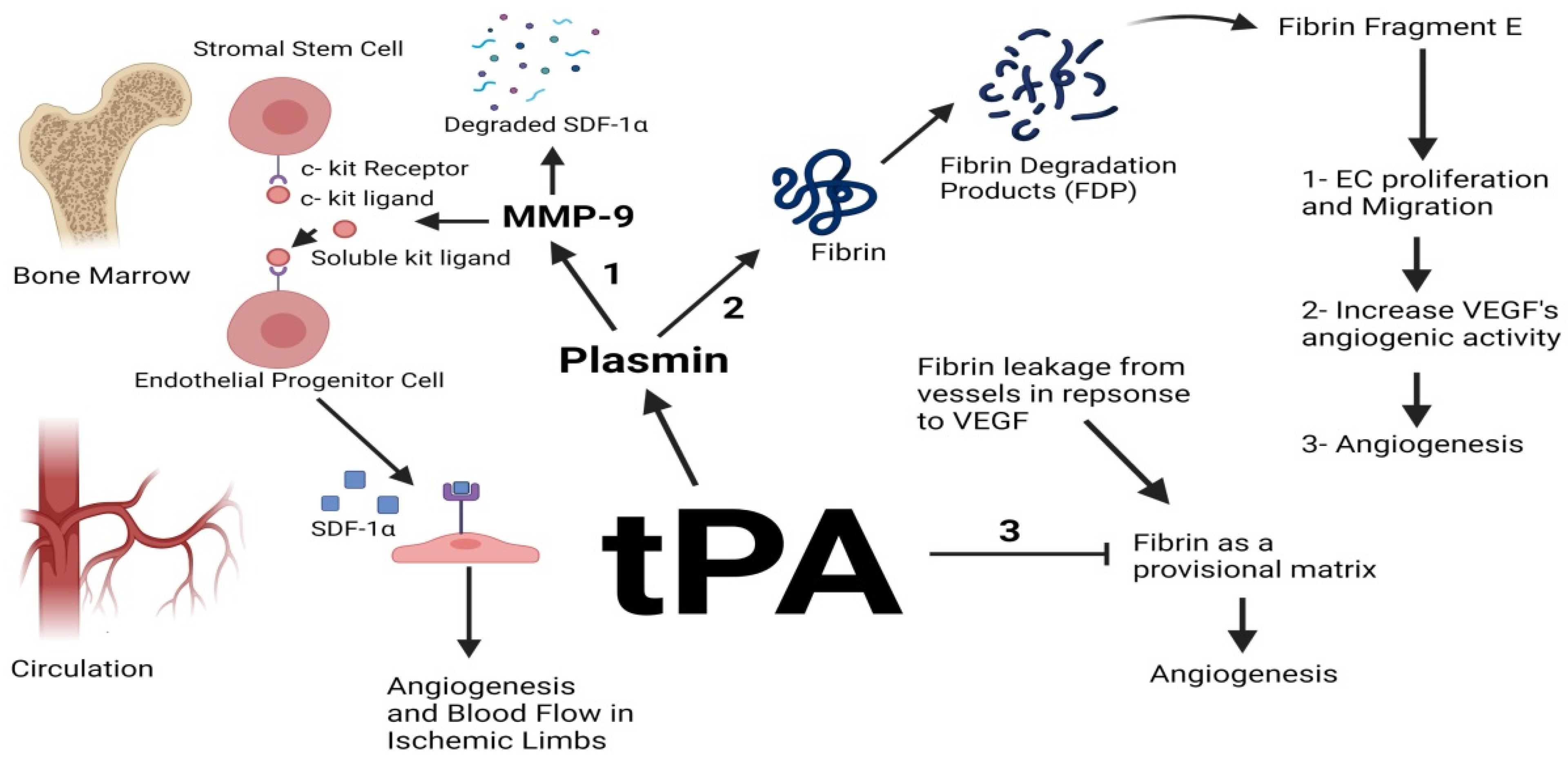
2.1. tPA
2.2. uPA

{kind=link}
{kind=link}
{kind=link}
| Model | Outcome | Mechanism |
|---|---|---|
| uPA and uPAR deficient mice implanted with murine prostate cancer cells |
| Reduced tumor size in uPA and uPAR deficient mice could be due to the reduction of macrophage number [49] |
| Stable transfection of SNB19 cells with antisense-uPA |
| uPa deficiency decreased PI3K and Akt phosphorylation and actin cytoskeleton formation [50] |
| uPA deficient mice implanted with malignant murine keratinocytes |
| uPA deficiency was recompensed by tPA [51] |
| MCF-7 cells treated with single-chained uPA (scuPA) and uPA amino-terminal fragment (ATF) |
| scuPA and uPA ATF induced the Phosphorylation and activation of ERK1/2 [57] |
2.3. uPAR
| Model | Outcome | Mechanism |
|---|---|---|
| HUVEC cells incubated with tumor conditioned media | Enhanced EC invasion and migration | Soluble uPAR from the tumor conditioned media colocalized in membrane lipid rafts on EC and induced ERK/Rac-1 mediated cellular migration and tube formation [69] |
|
| Upon VEGF stimulation, uPAR and integrins interact and are endocytosed via a clathrin-coated vesicle followed by their redistribution to the leading edge of the cell to focus the proteolytic activity of plasmin at the invading side of the cell [64] |
|
| High levels of uPAR lead to increased levels of integrins and enhanced adhesion to fibronectin, thus fibronectin-dependent activation of ERK and stimulation of cellular proliferation [62] |
| HUVEC cells transfected with uPAR small-interfering RNA; subsequent VEGF treatment |
| VEGF prompts the interaction of VEGFR2 with uPAR; uPAR then induces the endocytosis of the complex and the activation of VEGFR2 signaling [66] |
2.4. Plasminogen/Plasmin
2.5. PAI-1
2.5.1. PAI-1 Promotes Angiogenesis through Interacting with Proteases
2.5.2. PAI-1 Promotes Angiogenesis through Binding to Vitronectin
2.5.3. PAI-1 Promotes Angiogenesis through Inhibition of Apoptosis
| Model | Outcome | Mechanism |
|---|---|---|
|
| In the absence of TGF-β, an increase in miR-30′s expression causes a decrease in PAI-1′s expression and the subsequent accumulation of plasmin which instigates the degradation of fibrin and the inhibition of angiogenesis [94] |
|
| Knockdown of PAI-1 enhances plasmin activity which cleaves Fas ligand and releases it as a soluble 21.5 kDa soluble protein with proapoptotic properties [105] |
| Adenovirus-mediated gene transfer of mutated PAI-1 (PAI-1 deficient in vitronectin binding or in plasminogen activators inhibition) to PAI-1 deficient mice transplanted with malignant murine keratinocytes | Restoration of tumor angiogenesis with recombinant PAI-1 mutated at the vitronectin interaction site | PAI-1 induces tumor angiogenesis and invasion through its interaction with proteases, not vitronectin [51] |
2.5.4. PAI-1 in Vessel Attrition
2.6. PAI-2
3. Antiangiogenic Therapy: Targeting Serine Proteases
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adair, T.H.; Montani, J.-P. Angiogenesis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010; pp. 1–13. [Google Scholar]
- Lammert, E.; Axnick, J. Vascular Lumen Formation. Cold Spring Harb. Perspect. Med. 2011, 2, a006619. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Wagner, N.; Wagner, K.-D. The Emerging Role of PPAR Beta/Delta in Tumor Angiogenesis. PPAR Res. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Yoshitomi, Y.; Ikeda, T.; Saito-Takatsuji, H.; Yonekura, H. Emerging Role of AP-1 Transcription Factor JunB in Angiogenesis and Vascular Development. Int. J. Mol. Sci. 2021, 22, 2804. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Polverini, P. The Pathophysiology of Angiogenesis. Crit. Rev. Oral Biol. Med. 1995, 6, 230–247. [Google Scholar] [CrossRef]
- Chen, X.; Man, G.C.W.; Liu, Y.; Wu, F.; Huang, J.; Li, T.C.; Wang, C.C. Physiological and pathological angiogenesis in endometrium at the time of embryo implantation. Am. J. Reprod. Immunol. 2017, 78, e12693. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Webster, K.; Bhatt, A.; Tian, H.; Su, G.; Li, W. Concurrent Physiological and Pathological Angiogenesis in Retinopathy of Prematurity and Emerging Therapies. Int. J. Mol. Sci. 2021, 22, 4809. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S. Role of the Matrix Metalloproteinase and Plasminogen Activator–Plasmin Systems in Angiogenesis. Arter. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Belinha, J.; Jorge, R.N. Modelling skin wound healing angiogenesis: A review. J. Theor. Biol. 2018, 459, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. History of Angiogenesis. In Angiogenesis; Springer: Singapore, 2008; pp. 1–14. [Google Scholar]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nat. Cell Biol. 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in Cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Flegg, J.A.; Menon, S.N.; Byrne, H.M.; McElwain, D.L.S. A Current Perspective on Wound Healing and Tumour-Induced Angiogenesis. Bull. Math. Biol. 2020, 82, 23. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)-key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Ziyad, S.; Iruela-Arispe, M.L. Molecular Mechanisms of Tumor Angiogenesis. Genes Cancer 2011, 2, 1085–1096. [Google Scholar] [CrossRef] [Green Version]
- Bajou, K.; Declerck, Y.; Laug, W. Proteinases and Their Inhibitors in Angiogenesis. In Antiangiogenic Cancer Therapy; Taylor and Francis Group: Boca Raton, FL, USA, 2007; pp. 239–250. [Google Scholar]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Masson, V.; de la Ballina, L.R.; Munaut, C.; Wielockx, B.; Jost, M.; Maillard, C.; Blacher, S.; Bajou, K.; Itoh, T.; Itohara, S.; et al. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 2004, 19, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rakic, J.M.; Maillard, C.; Jost, M.; Bajou, K.; Masson, V.; Devy, L.; Lambert, V.; Foidart, J.M. Role of plasminogen activator-plasmin system in tumor angiogenesis. Cell. Mol. Life Sci. 2003, 60, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.-W.; Hoover-Plow, J.; Plow, E.F. The role of plasminogen in angiogenesis in vivo. J. Thromb. Haemost. 2003, 1, 1683–1687. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Holloway, R.; Miller, V.; Waisman, D. Plasmin and Plasminogen System in the Tumor Microenvironment: Implications for Cancer Diagnosis, Prognosis, and Therapy. Cancers 2021, 13, 1838. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef] [Green Version]
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 622473. [Google Scholar] [CrossRef] [PubMed]
- Schaller, J.; Gerber, S.S. The plasmin–antiplasmin system: Structural and functional aspects. Cell. Mol. Life Sci. 2010, 68, 785–801. [Google Scholar] [CrossRef] [Green Version]
- Chevilley, A.; Lesept, F.; Lenoir, S.; Ali, C.; Parcq, J.; Vivien, D. Impacts of tissue-type plasminogen activator (tPA) on neuronal survival. Front. Cell. Neurosci. 2015, 9, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakic, J.-M.; Lambert, V.; Munaut, C.; Bajou, K.; Peyrollier, K.; Alvarez-Gonzalez, M.-L.; Carmeliet, P.; Foidart, J.-M.; Noel, A. Mice without uPA, tPA, or Plasminogen Genes Are Resistant to Experimental Choroidal Neovascularization. Investig. Opthalmol. Vis. Sci. 2003, 44, 1732–1739. [Google Scholar] [CrossRef]
- Brodsky, S.; Chen, J.; Lee, A.; Akassoglou, K.; Norman, J.; Goligorsky, M.S. Plasmin-dependent and -independent effects of plasminogen activators and inhibitor-1 on ex vivo angiogenesis. Am. J. Physiol. Circ. Physiol. 2001, 281, H1784–H1792. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.; Day, Y.-J.; Sun, C.-K.; Yip, H.-K. TPA-MMP-9 Axis Plays a Pivotal Role in Mobilization of Endothelial Progenitor Cells from Bone Marrow to Circulation and Ischemic Region for Angiogenesis. Stem Cells Int. 2016, 2016, 5417565. [Google Scholar] [CrossRef] [Green Version]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.; et al. Recruitment of Stem and Progenitor Cells from the Bone Marrow Niche Requires MMP-9 Mediated Release of Kit-Ligand. Cell 2002, 109, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Leu, S.; Lu, H.-I.; Sun, C.-K.; Sheu, J.-J.; Chen, Y.-L.; Tsai, T.-H.; Yeh, K.-H.; Chai, H.-T.; Chua, S.; Tsai, C.-Y.; et al. Retention of endothelial progenitor cells in bone marrow in a murine model of endogenous tissue plasminogen activator (tPA) deficiency in response to critical limb ischemia. Int. J. Cardiol. 2014, 170, 394–405. [Google Scholar] [CrossRef]
- Prager, G.W.; Breuss, J.M.; Steurer, S.; Olcaydu, D.; Mihaly, J.; Brunner, P.M.; Stockinger, H.; Binder, B.R. Vascular Endothelial Growth Factor Receptor-2–Induced Initial Endothelial Cell Migration Depends on the Presence of the Urokinase Receptor. Circ. Res. 2004, 94, 1562–1570. [Google Scholar] [CrossRef] [Green Version]
- Thompson, W.D.; Smith, E.B.; Stirk, C.M.; Marshall, F.I.; Stout, A.J.; Kocchar, A. Angiogenic activity of fibrin degradation products is located in fibrin fragment E. J. Pathol. 1992, 168, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bootle-Wilbraham, C.A.; Tazzyman, S.; Thompson, W.D.; Stirk, C.M.; Lewis, C.E. Fibrin Fragment E stimulates he proliferation, migration, and differentiation of human microvascular endothelial cells in vitro. Angiogenesis 2001, 4, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Stirk, C.M.; Smith, E.B.; Thompson, W. Smooth Muscle Cell Outgrowth Stimulated by Fibrin Degradation Products: The Potential Role of Fibrin Fragment E in Restenosis and Atherogenesis. Thromb. Res. 2000, 98, 165–174. [Google Scholar] [CrossRef]
- Arai, K.-I.; Yasukawa, T.; Kato, A.; Kubota, A.; Usui, H.; Takase, N.; Kuwayama, S.; Ogura, Y. Tissue Plasminogen Activator as an Antiangiogenic Agent in Experimental Corneal Neovascularization in Rabbits. Ophthalmic Res. 2018, 59, 170–175. [Google Scholar] [CrossRef]
- Sun, Q.; Shen, Y.; Su, L.; Xu, X. Inhibition of Pathological Retinal Neovascularization by a Small Peptide Derived from Human Tissue-Type Plasminogen Kringle 2. Front. Pharmacol. 2020, 10, 1639. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Klijn, J.G.; Henzen-Logmand, S.C.; Bouwman, I.; Van Roozendaal, K.E.; Peters, H.A.; Setyono-Han, B.; Foekens, J.A. Urokinase-Type-Plasminogen-Activator (UPA) Production by Human Breast (Myo) Fibroblasts in Vitro: Influence of Transforming Growth Factor-Beta(1) (TGF Beta(1)) Compared with Factor(s) Released by Human Epithelial-Carcinoma Cells. Int. J. Cancer 1998, 76, 829–835. [Google Scholar] [CrossRef]
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noël, A.; Foidart, J.-M. Role of Serine Proteases and their Inhibitors in Tumor Growth and Angiogenesis. In Lymphangiogenesis in Cancer Metastasis; Springer: Singapore, 2002; Volume 4, pp. 23–38. [Google Scholar]
- Carmeliet, P.; Moons, L.; Herbert, J.-M.; Crawley, J.; Lupu, F.; Lijnen, R.; Collen, D. Urokinase but Not Tissue Plasminogen Activator Mediates Arterial Neointima Formation in Mice. Circ. Res. 1997, 81, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, S.; McMahon, G.; Petitclerc, E.; Lawrence, D. Plasminogen Activator Inhibitor-1 in Tumor Growth, Angiogenesis and Vascular Remodeling. Curr. Pharm. Des. 2003, 9, 1545–1564. [Google Scholar] [CrossRef]
- Bacharach, E.; Itin, A.; Keshet, E. In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc. Natl. Acad. Sci. USA 1992, 89, 10686–10690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymans, S.; Luttun, A.; Nuyens, D.; Theilmeier, G.; Creemers, E.; Moons, L.; Dyspersin, G.; Cleutjens, J.; Shipley, M.; Angellilo, A.; et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999, 5, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sud, S.; Mizutani, K.; Gyetko, M.R.; Pienta, K.J. Activation of Urokinase Plasminogen Activator and Its Receptor Axis Is Essential for Macrophage Infiltration in a Prostate Cancer Mouse Model. Neoplasia 2011, 13, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, N.; Mohanam, S.; Gujrati, M.; Olivero, W.C.; Dinh, D.H.; Rao, J.S. Downregulation of uPA inhibits migration and PI3k/Akt signaling in glioblastoma cells. Oncogene 2003, 22, 392–400. [Google Scholar] [CrossRef]
- Bajou, K.; Masson, V.; Gerard, R.D.; Schmitt, P.M.; Albert, V.; Praus, M.; Lund, L.R.; Frandsen, T.L.; Brunner, N.; Dano, K.; et al. The Plasminogen Activator Inhibitor PAI-1 Controls in Vivo Tumor Vascularization by Interaction with Proteases, Not Vitronectin. J. Cell Biol. 2001, 152, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kenny, H.A.; Swindell, E.P.; Mitra, A.K.; Hankins, P.L.; Ahn, R.W.; Gwin, K.; Mazar, A.P.; O’Halloran, T.V.; Lengyel, E. Urokinase Plasminogen Activator System–Targeted Delivery of Nanobins as a Novel Ovarian Cancer Therapy. Mol. Cancer Ther. 2013, 12, 2628–2639. [Google Scholar] [CrossRef] [Green Version]
- Devy, L.; Blacher, S.; Grignet-Debrus, C.; Bajou, K.; Masson, V.; Gerard, R.D.; Gils, A.; Carmeliet, G.; Carmeliet, P.; Declerck, P.J.; et al. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 2002, 16, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Rubina, K.A.; Sysoeva, V.; Zagorujko, E.I.; Tsokolaeva, Z.I.; Kurdina, M.I.; Parfyonova, Y.; Tkachuk, V.A. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch. Dermatol. Res. 2017, 309, 433–442. [Google Scholar] [CrossRef]
- Banys-Paluchowski, M.; Witzel, I.; Aktas, B.; Fasching, P.A.; Hartkopf, A.; Janni, W.; Kasimir-Bauer, S.; Pantel, K.; Schön, G.; Rack, B.; et al. The prognostic relevance of urokinase-type plasminogen activator (uPA) in the blood of patients with metastatic breast cancer. Sci. Rep. 2019, 9, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, V.; Jayaraman, P.-S.; Zaitsev, S.V.; Lebedeva, T.; Bdeir, K.; Kershaw, R.; Holman, K.R.; Parfyonova, Y.; Semina, E.V.; Beloglazova, I.; et al. Urokinase-type Plasminogen Activator (uPA) Promotes Angiogenesis by Attenuating Proline-rich Homeodomain Protein (PRH) Transcription Factor Activity and De-repressing Vascular Endothelial Growth Factor (VEGF) Receptor Expression. J. Biol. Chem. 2016, 291, 15029–15045. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Hussaini, I.M.; Gonias, S.L. Binding of Urokinase-type Plasminogen Activator to Its Receptor in MCF-7 Cells Activates Extracellular Signal-regulated Kinase 1 and 2 Which Is Required for Increased Cellular Motility. J. Biol. Chem. 1998, 273, 8502–8507. [Google Scholar] [CrossRef] [Green Version]
- Montuori, N.; Ragno, P. Role of uPA/uPAR in the Modulation of Angiogenesis. Chem. Immunol. Allergy 2013, 99, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, L.; Aguirre-Ghiso, J.A. Urokinase receptor and integrin partnership: Coordination of signaling for cell adhesion, migration and growth. Curr. Opin. Cell Biol. 2000, 12, 613–620. [Google Scholar] [CrossRef]
- Breuss, J.M.; Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adhes. Migr. 2012, 6, 535–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugge, T.H.; Suh, T.T.; Flick, M.J.; Daugherty, C.C.; John, R.; Solberg, H.; Ellis, V.; Danø, K.; Degen, J.L. The Receptor for Urokinase-type Plasminogen Activator Is Not Essential for Mouse Development or Fertility. J. Biol. Chem. 1995, 270, 16886–16894. [Google Scholar] [CrossRef] [Green Version]
- Ghiso, J.A.A.; Kovalski, K.; Ossowski, L. Tumor Dormancy Induced by Downregulation of Urokinase Receptor in Human Carcinoma Involves Integrin and MAPK Signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Uhrin, P.; Breuss, J.M. UPAR: A Modulator of VEGF-Induced Angiogenesis: A Modulator of VEGF-Induced Angiogenesis. Cell Adh. Migr. 2013, 7, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Alexander, R.A.; Prager, G.W.; Mihaly-Bison, J.; Uhrin, P.; Sunzenauer, S.; Binder, B.R.; Schütz, G.J.; Freissmuth, M.; Breuss, J.M. VEGF-Induced Endothelial Cell Migration Requires Urokinase Receptor (UPAR)-Dependent Integrin Redistribution. Cardiovasc. Res. 2012, 94, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herkenne, S.; Paques, C.; Nivelles, O.; Lion, M.; Bajou, K.; Pollenus, T.; Fontaine, M.; Carmeliet, P.; Martial, J.A.; Nguyen, N.-Q.-N.; et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis. Sci. Signal. 2015, 8, ra117. [Google Scholar] [CrossRef]
- Erkut, N.; Menteşe, A.; Özbaş, H.M.; Ermantaş, N.; Sümer, A.; Örem, A.; Sönmez, M. The Prognostic Significance of Soluble Urokinase Plasminogen Activator Receptor in Acute Myeloid Leukemia. Turk. J. Hematol. 2016, 33, 135–140. [Google Scholar] [CrossRef]
- Rasmussen, L.J.; Schultz, M.; Iversen, K.; Eugen-Olsen, J.; Helms, M.; David, K.; Kjaer, A.; Lebech, A.-M.; Kronborg, G. Soluble urokinase plasminogen activator receptor (suPAR) is lower in disease-free patients but cannot rule out incident disease in patients with suspected cancer. Clin. Biochem. 2020, 84, 31–37. [Google Scholar] [CrossRef]
- Rao, J.S.; Gujrati, M.; Chetty, C. Tumor-Associated Soluble UPAR-Directed Endothelial Cell Motility and Tumor Angiogenesis. Oncogenesis 2013, 2, e53. [Google Scholar] [CrossRef] [Green Version]
- Min, H.Y.; Doyle, L.V.; Vitt, C.R.; Zandonella, C.; Stratton-Thomas, J.R.; A Shuman, M.; Rosenberg, S. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Res. 1996, 56, 2428–2433. [Google Scholar] [PubMed]
- Li, H.; Lu, H.; Griscelli, F.; Opolon, P.; Sun, L.-Q.; Ragot, T.; Legrand, Y.; Belin, D.; Soria, J.; Soria, C.; et al. Adenovirus-mediated delivery of a uPA/uPAR antagonist suppresses angiogenesis-dependent tumor growth and dissemination in mice. Gene Ther. 1998, 5, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannocks, M.J.; Oliver, L.; Gabrilove, J.L.; Wilson, E.L. Regulation of Proteolytic Activity in Human Bone Marrow Stromal Cells by Basic Fibroblast Growth Factor, Interleukin-1, and Transforming Growth Factor Beta. Blood 1992, 79, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Noel, A.; Gilles, C.; Bajou, K.; Devy, L.; Kebers, F.; Lewalle, J.M.; Maquoi, E.; Munaut, C.; Remacle, A.; Foidart, J.M. Emerging roles for proteinases in cancer. Invasion Metastasis 1997, 17, 221–239. [Google Scholar]
- Napolitano, F.; Montuori, N. The Role of the Plasminogen Activation System in Angioedema: Novel Insights on the Pathogenesis. J. Clin. Med. 2021, 10, 518. [Google Scholar] [CrossRef]
- Baramova, E.; Bajou, K.; Remacle, A.; L’Hoir, C.; Krell, H.; Weidle, U.; Noel, A.; Foidart, J. Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett. 1997, 405, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Chopp, M.; Xin, H.; Zhang, L.; Wang, F.; Golembieski, W.; Zhang, Z.G.; He, L.; Liu, Z. Plasminogen deficiency causes reduced angiogenesis and behavioral recovery after stroke in mice. Br. J. Pharmacol. 2021, 271678, 211007958. [Google Scholar] [CrossRef] [PubMed]
- Künnapuu, J.; Bokharaie, H.; Jeltsch, M. Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs. Biology 2021, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-S.; Svenningsson, P. Modulation of Ion Channels and Receptors by p11 (S100A10). Trends Pharmacol. Sci. 2020, 41, 487–497. [Google Scholar] [CrossRef]
- Madureira, P.; O’Connell, P.A.; Surette, A.P.; Miller, V.A.; Waisman, D.M. The Biochemistry and Regulation of S100A10: A Multifunctional Plasminogen Receptor Involved in Oncogenesis. J. Biomed. Biotechnol. 2012, 2012, 353687. [Google Scholar] [CrossRef] [Green Version]
- Surette, A.P.; Madureira, P.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; He, Z.; Lin, M.; Wang, Y.; Yang, F.; Mitteer, R.A.; Kim, H.-J.; Yeo, E.; Han, H.; Qin, L.; et al. Plasminogen regulates mesenchymal stem cell–mediated tissue repair after ischemia through Cyr61 activation. JCI Insight 2020, 5, e131376. [Google Scholar] [CrossRef]
- Yamamoto, H.; Okada, R.; Iguchi, K.; Ohno, S.; Yokogawa, T.; Nishikawa, K.; Unno, K.; Hoshino, M.; Takeda, A. Involvement of plasmin-mediated extracellular activation of progalanin in angiogenesis. Biochem. Biophys. Res. Commun. 2013, 430, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xue, L. Angiostatin. Semin. Thromb. Hemost. 2004, 30, 83–93. [Google Scholar] [CrossRef]
- Loskutoff, D.J.; van Mourik, J.A.; Erickson, L.A.; Lawrence, D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2956–2960. [Google Scholar] [CrossRef] [Green Version]
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen activator inhibitor 1: Physiological and pathophysiological roles. News Physiol. Sci. 2002, 17, 56–61. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2011, 227, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Kubala, M.H.; Declerck, Y.A. The plasminogen activator inhibitor-1 paradox in cancer: A mechanistic understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef]
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066. [Google Scholar] [CrossRef] [PubMed]
- Loskutoff, D.J.; Curriden, S.A.; Hu, G.; Deng, G. Regulation of cell adhesion by PAI-1. APMIS J. Pathol. Microbiol. Immunol. 1999, 107, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.-P.; Wilkins-Port, C.E.; Higgins, S.P.; Freytag, J.; Overstreet, J.M.; Klein, R.M.; Higgins, C.E.; Samarakoon, R.; Higgins, P.J. PAI-1: An Integrator of Cell Signaling and Migration. Int. J. Cell Biol. 2011, 2011, 562481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Gils, A.; Declerck, P.J. Plasminogen Activator Inhibitor-1. Curr. Med. Chem. 2004, 11, 2323–2334. [Google Scholar] [CrossRef]
- Pinsky, D.J.; Liao, H.; A Lawson, C.; Yan, S.F.; Chen, J.; Carmeliet, P.; Loskutoff, D.J.; Stern, D.M. Coordinated induction of plasminogen activator inhibitor-1 (PAI-1) and inhibition of plasminogen activator gene expression by hypoxia promotes pulmonary vascular fibrin deposition. J. Clin. Investig. 1998, 102, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, J.V.; Xiao, L.; Kim, D.J.; Khan, O.F.; Kowalski, P.S.; Anderson, D.G.; Pecot, C.V.; Azam, S.H.; Parker, J.S.; Tsai, Y.S.; et al. Endothelial miR-30c suppresses tumor growth via inhibition of TGF-β–induced Serpine1. J. Clin. Investig. 2019, 129, 1654–1670. [Google Scholar] [CrossRef]
- Pepper, M.; Montesano, R. Proteolytic balance and capillary morphogenesis. Cell Differ. Dev. 1990, 32, 319–327. [Google Scholar] [CrossRef]
- Kwak, T.J.; Lee, E. In vitro modeling of solid tumor interactions with perfused blood vessels. Sci. Rep. 2020, 10, 20142. [Google Scholar] [CrossRef]
- Chu, Y.; Bucci, J.C.; Peterson, C.B. Identification of a PAI-1-binding site within an intrinsically disordered region of vitronectin. Protein Sci. 2020, 29, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Schroeck, F.; De Prada, N.A.; Sperl, S.; Schmitt, M.; Magdolen, V. Interaction of Plasminogen Activator Inhibitor Type-1 (PAI-1) with Vitronectin (Vn): Mapping the Binding Sites on PAI-1 and Vn. Biol. Chem. 2002, 383, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Huang, T.-S. Plasminogen activator inhibitor-1: The expression, biological functions, and effects on tumorigenesis and tumor cell adhesion and migration. J. Cancer Mol. 2005, 1, 25–36. [Google Scholar]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of uPAR promotes urokinase translocation into the nucleus and epithelial to mesenchymal transition in neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef] [PubMed]
- Isogai, C.; E Laug, W.; Shimada, H.; Declerck, P.; Stins, M.F.; Durden, D.L.; Epstein, A.; DeClerck, Y. Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res. 2001, 61, 5587–5594. [Google Scholar] [PubMed]
- Takahashi, T.; Suzuki, K.; Ihara, H.; Mogami, H.; Kazui, T.; Urano, T. Plasminogen Activator Inhibitor Type 1 Promotes Fibrosarcoma Cell Migration by Modifying Cellular Attachment to Vitronectin via Alpha(v)Beta(5) Integrin. Semin. Thromb. Hemost. 2005, 31, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Bajou, K.; Peng, H.; Laug, W.E.; Maillard, C.; Noel, A.; Foidart, J.M.; Martial, J.A.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 Protects Endothelial Cells from FasL-Mediated Apoptosis. Cancer Cell 2008, 14, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Placencio, V.R.; Declerck, Y.A. Protumorigenic Activity of Plasminogen Activator Inhibitor-1 Through an Antiapoptotic Function. J. Natl. Cancer Inst. 2012, 104, 1470–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajou, K.; Noël, A.; Gerard, R.; Masson, V.; Brunner, N.; Holst-Hansen, C.; Skobe, M.; Fusenig, N.; Carmeliet, P.; Collen, D.; et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat. Med. 1998, 4, 923–928. [Google Scholar] [CrossRef]
- Fortenberry, Y.M.; Brandal, S.M.; Carpentier, G.; Hemani, M.; Pathak, A.P. Intracellular Expression of PAI-1 Specific Aptamers Alters Breast Cancer Cell Migration, Invasion and Angiogenesis. PLoS ONE 2016, 11, e0164288. [Google Scholar] [CrossRef] [PubMed]
- Bajou, K.; Herkenne, S.; Thijssen, V.L.; D’Amico, S.; Nguyen, N.-Q.-N.; Bouché, A.; Tabruyn, S.; Srahna, M.; Carabin, J.-Y.; Nivelles, O.; et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat. Med. 2014, 20, 741–747. [Google Scholar] [CrossRef]
- Lambert, V.; Munaut, C.; Noël, A.; Frankenne, F.; Bajou, K.; Gerard, R.; Carmeliet, P.; Defresne, M.P.; Foidart, J.M.; Rakic, J.M. Influence of Plasminogen Activator Inhibitor Type 1 on Choroidal Neovascularization. FASEB J. 2001, 15, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajou, K.; Maillard, C.; Jost, M.; Lijnen, R.H.; Gils, A.; Declerck, P.; Carmeliet, P.; Foidart, J.-M.; Noel, A. Host-Derived Plas-minogen Activator Inhibitor-1 (PAI-1) Concentration Is Critical for in Vivo Tumoral Angiogenesis and Growth. Oncogene 2004, 23, 6986–6990. [Google Scholar] [CrossRef] [Green Version]
- Eitzman, D.T.; Krauss, J.C.; Shen, T.; Cui, J. Ginsburg Lack of plasminogen activator inhibitor-1 effect in a transgenic mouse model of metastatic melanoma. Blood 1996, 87, 4718–4722. [Google Scholar] [PubMed]
- Soff, G.A.; Sanderowitz, J.; Gately, S.; Verrusio, E.; Weiss, I.; Brem, S.; Kwaan, H.C. Expression of plasminogen activator inhibitor type 1 by human prostate carcinoma cells inhibits primary tumor growth, tumor-associated angiogenesis, and metastasis to lung and liver in an athymic mouse model. J. Clin. Investig. 1995, 96, 2593–2600. [Google Scholar] [CrossRef]
- Takeshita, K.; Yamamoto, K.; Ito, M.; Kondo, T.; Matsushita, T.; Hirai, M.; Kojima, T.; Nishimura, M.; Nabeshima, Y.; Loskutoff, D.J.; et al. Increased Expression of Plasminogen Activator Inhibitor-1 with Fibrin Deposition in a Murine Model of Aging, “Klotho” Mouse. Semin. Thromb. Hemost. 2002, 28, 545–554. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and Plasminogen Activator Inhibitor-1 (PAI-1) Regu-lation: Implication in the Pathogenesis of Thrombotic Disorders in the Elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Budinger, G.R.S.; Mutlu, G.M.; et al. PAI-1-regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7090–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci. Adv. 2017, 3, eaao1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sivan, U.; Tan, S.L.; Lippo, L.; De Angelis, J.; Labella, R.; Singh, A.; Chatzis, A.; Cheuk, S.; Medhghalchi, M.; et al. High-resolution 3D imaging uncovers organ-specific vascular control of tissue aging. Sci. Adv. 2021, 7, eabd7819. [Google Scholar] [CrossRef]
- Chen, J.; Lippo, L.; Labella, R.; Tan, S.L.; Marsden, B.D.; Dustin, M.L.; Ramasamy, S.K.; Kusumbe, A.P. Decreased blood vessel density and endothelial cell subset dynamics during ageing of the endocrine system. EMBO J. 2021, 40, e105242. [Google Scholar] [CrossRef]
- Boncela, J.; Przygodzka, P.; Papiewska-Pająk, I.; Wyroba, E.; Cierniewski, C.S. Association of Plasminogen Activator Inhibitor Type 2 (PAI-2) with Proteasome within Endothelial Cells Activated with Inflammatory Stimuli. J. Biol. Chem. 2011, 286, 43164–43171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrick, R.J.; Røjkjaer, L.P.; Yang, A.Y.; Roh, M.H.; Siebert, A.E.; Ginsburg, D. Deficiency of Plasminogen Activator In-hibitor-2 Results in Accelerated Tumor Growth. J. Thromb. Haemost. 2020, 18, 2968–2975. [Google Scholar] [CrossRef] [PubMed]
- Croucher, D.R.; Saunders, D.N.; Lobov, S.; Ranson, M. Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Rev. Cancer 2008, 8, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A.; Yerbury, J.J.; Farrawell, N.; Shearer, R.F.; Constantinescu, P.; Hatters, D.M.; Schroder, W.A.; Suhrbier, A.; Wilson, M.R.; Saunders, D.N.; et al. SerpinB2 (PAI-2) Modulates Proteostasis via Binding Misfolded Proteins and Promo-tion of Cytoprotective Inclusion Formation. PLoS ONE 2015, 10, e0130136. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Kim, H.S.; Choi, S.K.; Hwang, E.H.; Woo, J.; Ryu, H.S.; Kim, K.; Moon, A.; Moon, W.K. microRNA-200c/141 upregulates SerpinB2 to promote breast cancer cell metastasis and reduce patient survival. Oncotarget 2017, 8, 32769–32782. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Zhang, R.; Wang, Y.; Zhu, G.; Jin, T.; Li, C.; Zhang, S. MiR-200c/PAI-2 Promotes the Progression of Triple Negative Breast Cancer via M1/M2 Polarization Induction of Macrophage. Int. Immunopharmacol. 2020, 81, 106028. [Google Scholar] [CrossRef]
- Furuya, H.; Hayashi, K.; Shimizu, Y.; Kim, N.; Tsukikawa, Y.; Chen, R.; Sun, Y.; Chan, O.T.M.; Pagano, I.; Peres, R.; et al. Plasminogen activator inhibitor-2 (PAI-2) overexpression supports bladder cancer development in PAI-1 knockout mice in N-butyl-N- (4-hydroxybutyl)-nitrosamine- induced bladder cancer mouse model. J. Transl. Med. 2020, 18, 57. [Google Scholar] [CrossRef]
- Mack, G.S.; Marshall, A. Lost in migration. Nat. Biotechnol. 2010, 28, 214–229. [Google Scholar] [CrossRef]
- Meyer, J.E.; Brocks, C.; Graefe, H.; Mala, C.; Thäns, N.; Bürgle, M.; Rempel, A.; Rotter, N.; Wollenberg, B.; Lang, S. The Oral Serine Protease Inhibitor WX-671—First Experience in Patients with Advanced Head and Neck Carcinoma. Breast Care 2008, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Ertongur, S.; Lang, S.; Mack, B.; Wosikowski, K.; Muehlenweg, B.; Gires, O. Inhibition of the invasion capacity of carcinoma cells by WX-UK1, a novel synthetic inhibitor of the urokinase-type plasminogen activator system. Int. J. Cancer 2004, 110, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Setyono-Han, B.; Stürzebecher, J.; Schmalix, W.A.; Muehlenweg, B.; Sieuwerts, A.M.; Timmermans, M.; Magdolen, V.; Schmitt, M.; Klijn, J.G.M.; Foekens, J.A. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb. Haemost. 2005, 93, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Swiercz, R.; Skrzypczak-Jankun, E.; Merrell, M.M.; Selman, S.H.; Jankun, J. Angiostatic activity of synthetic inhibitors of urokinase type plasminogen activator. Oncol. Rep. 1999, 6, 523–529. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, Z.; An, J.; Mao, X.; Lin, F.; Sun, P. Effect of a synthetic inhibitor of urokinase plasminogen activator on the migration and invasion of human cervical cancer cells in vitro. Mol. Med. Rep. 2018, 17, 4273–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, B.; Majed, H.; Aboelela, A.; Minaei, E.; Jiang, L.; Fildes, K.; Cheung, C.-Y.; Johnson, D.; Bachovchin, D.; Cook, G.M.; et al. 6-Substituted amiloride derivatives as inhibitors of the urokinase-type plasminogen activator for use in metastatic disease. Bioorg. Med. Chem. Lett. 2019, 29, 126753. [Google Scholar] [CrossRef]
- Cai, J.; Ribkoff, J.; Olson, S.; Raghunathan, V.; Al-Samkari, H.; DeLoughery, T.G.; Shatzel, J.J. The many roles of tranexamic acid: An overview of the clinical indications for TXA in medical and surgical patients. Eur. J. Haematol. 2019, 104, 79–87. [Google Scholar] [CrossRef]
- Zhu, J.-W.; Ni, Y.-J.; Tong, X.-Y.; Guo, X.; Wu, X.-P.; Lu, Z.-F. Tranexamic Acid Inhibits Angiogenesis and Melanogenesis in Vitro by Targeting VEGF Receptors. Int. J. Med. Sci. 2020, 17, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Bala, H.R.; Lee, S.; Wong, C.; Pandya, A.; Rodrigues, M. Oral Tranexamic Acid for the Treatment of Melasma: A Review. Dermatol. Surg. 2018, 44, 814–825. [Google Scholar] [CrossRef]
- Yuan, C.; Guo, Z.; Yu, S.; Jiang, L.; Huang, M. Development of inhibitors for uPAR: Blocking the interaction of uPAR with its partners. Drug Discov. Today 2021, 26, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M.; Østergaard, S.; Gårdsvoll, H.; Kovalski, K.; Holst-Hansen, C.; Holm, A.; Ossowski, L.; Danø, K. Peptide-Derived Antagonists of the Urokinase Receptor. Affinity Maturation by Combinatorial Chemistry, Identification of Functional Epitopes, and Inhibitory Effect on Cancer Cell Intravasation. Biochemistry 2001, 40, 12157–12168. [Google Scholar] [CrossRef] [PubMed]
- Cammalleri, M.; Monte, M.D.; Locri, F.; Lista, L.; Aronsson, M.; Kvanta, A.; Rusciano, D.; De Rosa, M.; Pavone, V.; André, H.; et al. The Urokinase Receptor-Derived Peptide UPARANT Mitigates Angiogenesis in a Mouse Model of Laser-Induced Choroidal Neovascularization. Investig. Opthalmol. Vis. Sci. 2016, 57, 2600–2611. [Google Scholar] [CrossRef]
- Sillen, M.; Declerck, P. A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? Int. J. Mol. Sci. 2021, 22, 2721. [Google Scholar] [CrossRef] [PubMed]
- Izuhara, Y.; Takahashi, S.; Nangaku, M.; Takizawa, S.; Ishida, H.; Kurokawa, K.; van Ypersele de Strihou, C.; Hirayama, N.; Miyata, T. Inhibition of Plasminogen Activator Inhibitor-1: Its Mechanism and Effectiveness on Coagulation and Fibrosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 672–677. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, A.A.; Shaker, B.T.; Bajou, K. The Plasminogen–Activator Plasmin System in Physiological and Pathophysiological Angiogenesis. Int. J. Mol. Sci. 2022, 23, 337. https://doi.org/10.3390/ijms23010337
Ismail AA, Shaker BT, Bajou K. The Plasminogen–Activator Plasmin System in Physiological and Pathophysiological Angiogenesis. International Journal of Molecular Sciences. 2022; 23(1):337. https://doi.org/10.3390/ijms23010337
Chicago/Turabian StyleIsmail, Asmaa Anwar, Baraah Tariq Shaker, and Khalid Bajou. 2022. "The Plasminogen–Activator Plasmin System in Physiological and Pathophysiological Angiogenesis" International Journal of Molecular Sciences 23, no. 1: 337. https://doi.org/10.3390/ijms23010337
APA StyleIsmail, A. A., Shaker, B. T., & Bajou, K. (2022). The Plasminogen–Activator Plasmin System in Physiological and Pathophysiological Angiogenesis. International Journal of Molecular Sciences, 23(1), 337. https://doi.org/10.3390/ijms23010337