
Epigenetics of Cutaneous Sarcoma

Abstract
:1. Introduction
1.1. The Skin as an Exposure Site to Environmental Stimuli
1.2. The Skin Structure and Functions
1.3. Epigenetic Changes in the Skin
2. Dermatofibrosarcoma Protuberans (DFSP)
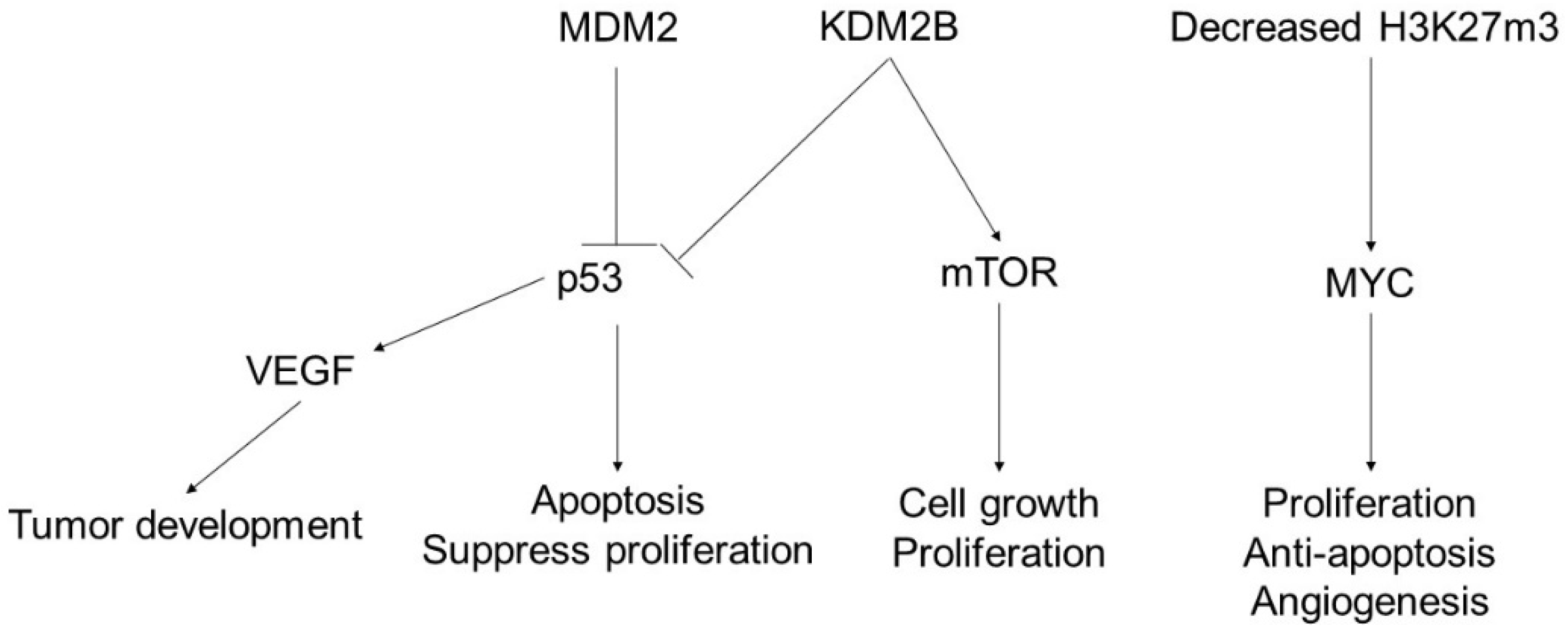
3. Angiosarcoma
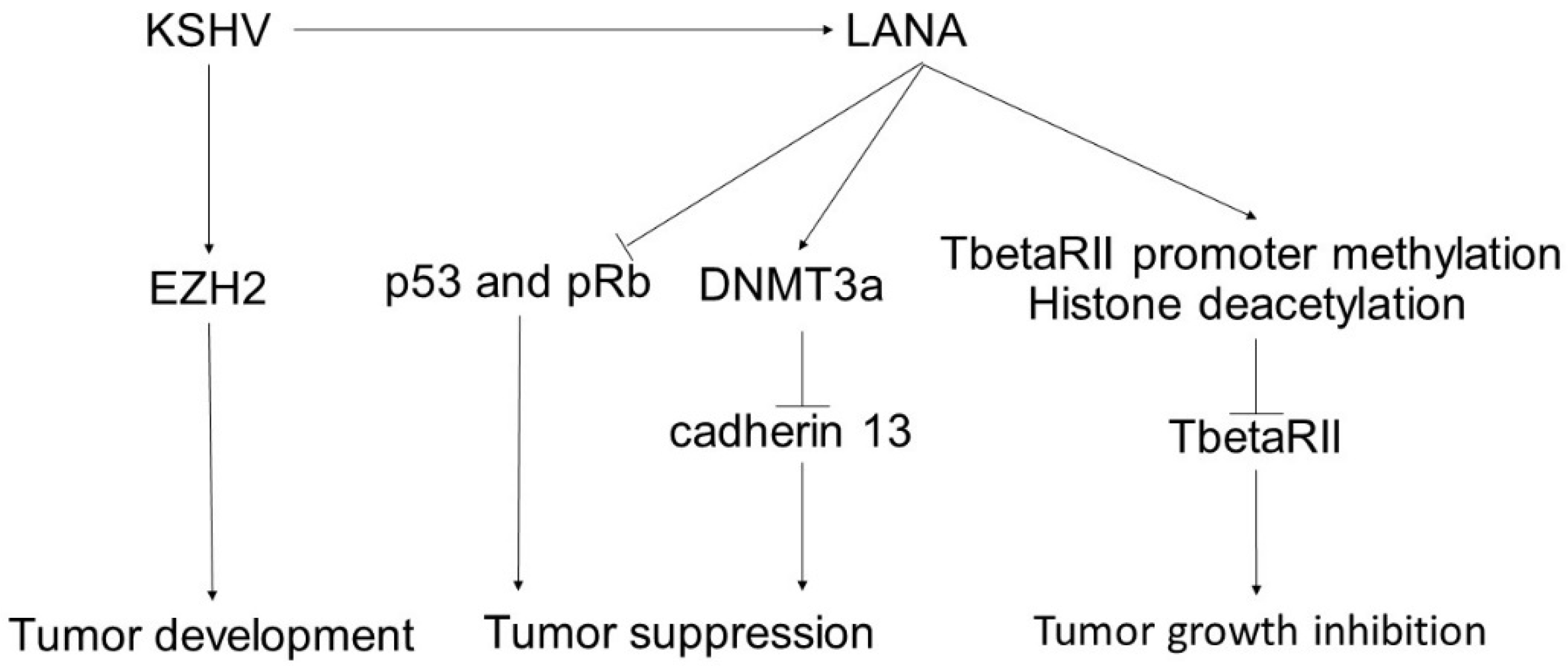
4. Kaposi’s Sarcoma
5. Leiomyosarcoma
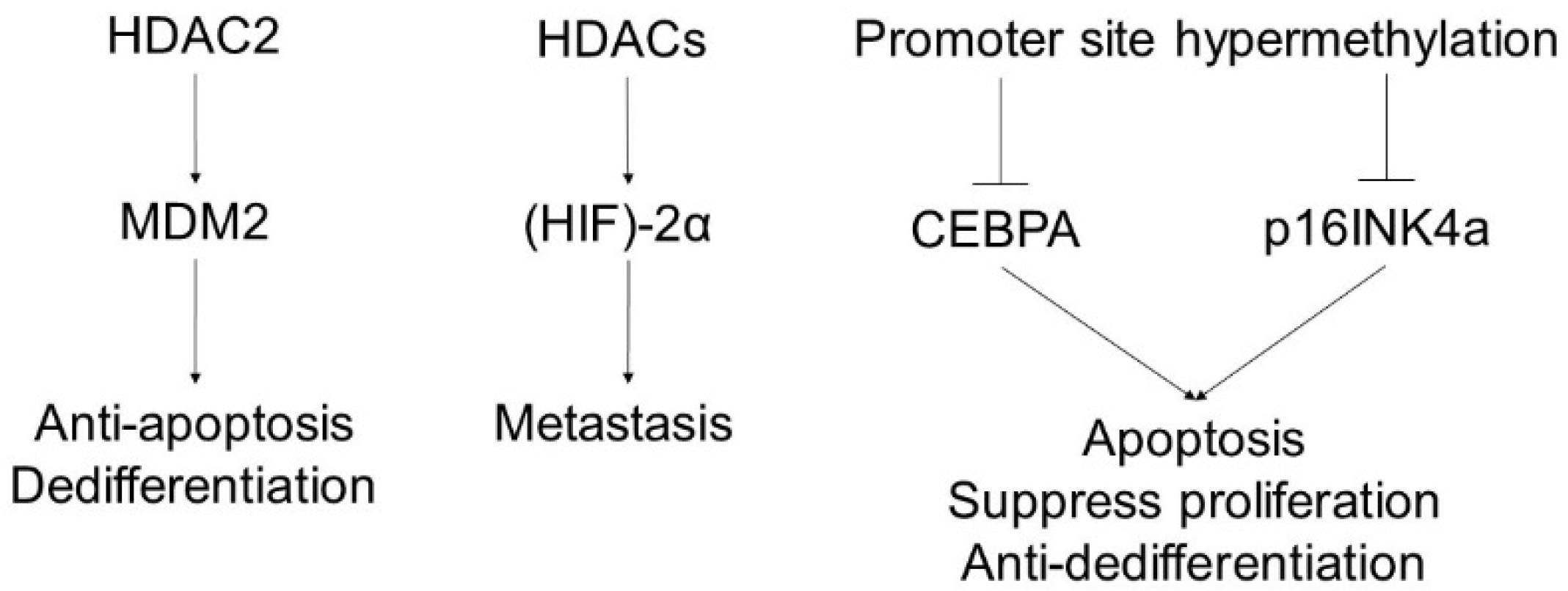
6. Liposarcoma
7. Radiotherapy-Induced Epigenetics Alteration
8. Summary of Epigenetic Modification in Cutaneous Sarcoma and Therapeutic Application
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.W.; Yeon, Y.; Han, J.Y.; Kim, E. Influence of exposure to summer environments on skin properties. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 2192–2196. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakatsuji, T.; Dokoshi, T.; Kulkarni, N.N.; Liggins, M.C.; Sen, G.; Gallo, R.L. Cutaneous innate immune tolerance is mediated by epigenetic control of MAP2K3 by HDAC8/9. Sci. Immunol. 2021, 6, 59. [Google Scholar] [CrossRef]
- Mervis, J.S.; McGee, J.S. DNA methylation and inflammatory skin diseases. Arch. Dermatol. Res. 2020, 312, 461–466. [Google Scholar] [CrossRef]
- Pastar, I.; Marjanovic, J.; Stone, R.C.; Chen, V.; Burgess, J.L.; Mervis, J.S.; Tomic-Canic, M. Epigenetic regulation of cellular functions in wound healing. Exp. Dermatol. 2021, 30, 1073–1089. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 8. [Google Scholar] [CrossRef]
- Kashyap, M.P.; Sinha, R.; Mukhtar, M.S.; Athar, M. Epigenetic regulation in the pathogenesis of non-melanoma skin cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Soboleva, A.G.; Mezentsev, A.; Zolotorenko, A.; Bruskin, S.; Pirusian, E. Three-dimensional skin models of psoriasis. Cells Tissues Organs 2014, 199, 301–310. [Google Scholar] [CrossRef]
- Sawada, Y.; Tong, Y.; Barangi, M.; Hata, T.; Williams, M.R.; Nakatsuji, T.; Gallo, R.L. Dilute bleach baths used for treatment of atopic dermatitis are not antimicrobial in vitro. J. Allergy Clin. Immunol. 2019, 143, 1946–1948. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E. Epithelial Skin Biology: Three Decades of Developmental Biology, a Hundred Questions Answered and a Thousand New Ones to Address. Curr. Top. Dev. Biol. 2016, 116, 357–374. [Google Scholar]
- Sakabe, J.; Kamiya, K.; Yamaguchi, H.; Ikeya, S.; Suzuki, T.; Aoshima, M.; Tatsuno, K.; Fujiyama, T.; Suzuki, M.; Yatagai, T.; et al. Proteome analysis of stratum corneum from atopic dermatitis patients by hybrid quadrupole-orbitrap mass spectrometer. J. Allergy Clin. Immunol. 2014, 134, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Honda, T.; Nakamizo, S.; Nakajima, S.; Nonomura, Y.; Otsuka, A.; Egawa, G.; Yoshimoto, T.; Nakamura, M.; Narumiya, S.; et al. Prostaglandin E(2) (PGE(2))-EP2 signaling negatively regulates murine atopic dermatitis-like skin inflammation by suppressing thymic stromal lymphopoietin expression. J. Allergy Clin. Immunol. 2019, 144, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M. Trial of Nemolizumab and Topical Agents for Atopic Dermatitis with Pruritus. N. Engl. J. Med. 2020, 383, 141–150. [Google Scholar] [CrossRef]
- Eichenfield, D.Z.; Sprague, J.; Eichenfield, L.F. Management of Acne Vulgaris: A Review. JAMA 2021, 326, 2055–2067. [Google Scholar] [CrossRef]
- Sawada, Y.; Gallo, R.L. Role of Epigenetics in the Regulation of Immune Functions of the Skin. J. Investig. Dermatol. 2021, 141, 1157–1166. [Google Scholar] [CrossRef]
- Beaziz, J.; Battistella, M.; Delyon, J.; Farges, C.; Marco, O.; Pages, C.; Le Maignan, C.; Da Meda, L.; Basset-Seguin, N.; Resche-Rigon, M.; et al. Long-Term Outcome of Neoadjuvant Tyrosine Kinase Inhibitors Followed by Complete Surgery in Locally Advanced Dermatofibrosarcoma Protuberans. Cancers 2021, 13, 9. [Google Scholar] [CrossRef]
- Cai, H.J.; Fang, J.H.; Cao, N.; Wang, W.; Kong, F.L.; Sun, X.X.; Huang, B. Dermatofibrosarcoma metastases to the pancreas: A case report. World J. Clin. Cases 2019, 7, 3316–3321. [Google Scholar] [CrossRef] [PubMed]
- Giacchero, D.; Maire, G.; Nuin, P.A.; Berthier, F.; Ebran, N.; Carlotti, A.; Celerier, P.; Coindre, J.M.; Esteve, E.; Fraitag, S.; et al. No correlation between the molecular subtype of COL1A1-PDGFB fusion gene and the clinico-histopathological features of dermatofibrosarcoma protuberans. J. Investig. Dermatol. 2010, 130, 904–907. [Google Scholar] [CrossRef] [Green Version]
- Criscione, V.D.; Weinstock, M.A. Descriptive epidemiology of dermatofibrosarcoma protuberans in the United States, 1973 to 2002. J. Am. Acad. Dermatol. 2007, 56, 968–973. [Google Scholar] [CrossRef]
- Rouhani, P.; Fletcher, C.D.; Devesa, S.S.; Toro, J.R. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: An analysis of 12,114 cases. Cancer 2008, 113, 616–627. [Google Scholar] [CrossRef]
- Protein CellMendenhall, W.M.; Zlotecki, R.A.; Scarborough, M.T. Dermatofibrosarcoma protuberans. Cancer 2004, 101, 2503–2508. [Google Scholar]
- Corvetta, D.; Chayka, O.; Gherardi, S.; D’Acunto, C.W.; Cantilena, S.; Valli, E.; Piotrowska, I.; Perini, G.; Sala, A. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J. Biol. Chem. 2013, 288, 8332–8341. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Cook, N.; Chen, J.; Zhou, J.; Wu, D. Embryonic Ectoderm Development (EED) as a Novel Target for Cancer Treatment. Curr. Top. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Martin, M.C.; Zeng, G.; Yu, J.; Schiltz, G.E. Small Molecule Approaches for Targeting the Polycomb Repressive Complex 2 (PRC2) in Cancer. J. Med. Chem. 2020, 63, 15344–15370. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, J.; Guo, Z.; Li, C.; Tan, Z.; Wang, J.; Yang, J.; Xue, L. Easy or Not-The Advances of EZH2 in Regulating T Cell Development, Differentiation, and Activation in Antitumor Immunity. Front. Immunol. 2021, 12, 741302. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Astolfi, A.; Gronchi, A.; Fontana, A.; Pantaleo, M.A.; Negri, T.; Brenca, M.; Tazzari, M.; Urbini, M.; Indio, V.; et al. Evolution of Dermatofibrosarcoma Protuberans to DFSP-Derived Fibrosarcoma: An Event Marked by Epithelial-Mesenchymal Transition-like Process and 22q Loss. Mol. Cancer Res. 2016, 14, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, N.; Macher-Goeppinger, S.; Pritsch, M.; Hüsing, J.; Hoppe-Seyler, K.; Schirmacher, P.; Pfitzenmaier, J.; Haferkamp, A.; Hoppe-Seyler, F.; Hohenfellner, M. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer 2010, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Yao, S.; Gomes, A.R.; Man, E.P.; Lee, H.J.; Gong, G.; Chang, S.; Kim, S.B.; Fujino, K.; Kim, S.W.; et al. BRCA1 positively regulates FOXO3 expression by restricting FOXO3 gene methylation and epigenetic silencing through targeting EZH2 in breast cancer. Oncogenesis 2016, 5, e214. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- van Kemenade, F.J.; Raaphorst, F.M.; Blokzijl, T.; Fieret, E.; Hamer, K.M.; Satijn, D.P.; Otte, A.P.; Meijer, C.J. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood 2001, 97, 3896–3901. [Google Scholar] [CrossRef] [Green Version]
- Takahira, T.; Oda, Y.; Tamiya, S.; Yamamoto, H.; Kawaguchi, K.; Kobayashi, C.; Oda, S.; Iwamoto, Y.; Tsuneyoshi, M. Microsatellite instability and p53 mutation associated with tumor progression in dermatofibrosarcoma protuberans. Hum. Pathol. 2004, 35, 240–245. [Google Scholar] [CrossRef]
- Khan, J.A.; Maki, R.G.; Ravi, V. Pathologic Angiogenesis of Malignant Vascular Sarcomas: Implications for Treatment. J. Clin. Oncol. 2018, 36, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; NS, I.J.; van der Graaf, W.T.A.; Kerst, J.M.; Schrage, Y.; Beijnen, J.H.; Steeghs, N.; van Houdt, W.J. Neoadjuvant Systemic Treatment of Primary Angiosarcoma. Cancers 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Coindre, J.M.; Terrier, P.; Guillou, L.; Le Doussal, V.; Collin, F.; Ranchère, D.; Sastre, X.; Vilain, M.O.; Bonichon, F.; N’Guyen Bui, B. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001, 91, 1914–1926. [Google Scholar] [PubMed]
- Zietz, C.; Rössle, M.; Haas, C.; Sendelhofert, A.; Hirschmann, A.; Stürzl, M.; Löhrs, U. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am. J. Pathol. 1998, 153, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Marion, M.J.; Lehman, T.; Welsh, J.; Harris, C.C.; Martel-Planche, G.; Kusters, I.; Montesano, R. p53 mutations at A:T base pairs in angiosarcomas of vinyl chloride-exposed factory workers. Carcinogenesis 1994, 15, 1–3. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; El-Naggar, A.K.; Xiong, S.; Yang, P.; Jackson, J.G.; Chau, G.; Lozano, G. Therapeutic efficacy of p53 restoration in Mdm2-overexpressing tumors. Mol. Cancer Res. 2014, 12, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Feller, J.K.; Mahalingam, M. c-myc and cutaneous vascular neoplasms. Am. J. Dermatopathol. 2013, 35, 364–369. [Google Scholar] [CrossRef]
- Italiano, A.; Chen, C.L.; Thomas, R.; Breen, M.; Bonnet, F.; Sevenet, N.; Longy, M.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: A pattern distinct from other sarcomas with complex genomics. Cancer 2012, 118, 5878–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, N.J.; Boguslawski, E.B.; Kuk, C.Y.; Chambers, C.M.; Duesbery, N.S. Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int. J. Oncol. 2015, 47, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Mentzel, T.; Kiss, K. Reduced H3K27me3 expression in radiation-associated angiosarcoma of the breast. Virchows Arch. 2018, 472, 361–368. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, S.; Vikram, A.; Hoffman, T.A.; Naqvi, A.; Lewarchik, C.M.; Kim, Y.R.; Irani, K. Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol. Arterioscler Thromb. Vasc. Biol. 2013, 33, 1936–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastow, R.; Mylne, J.S.; Lister, C.; Lippman, Z.; Martienssen, R.A.; Dean, C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 2004, 427, 164–167. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Kallin, E.M.; Tsukada, Y.; Zhang, Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat. Struct. Mol. Biol. 2008, 15, 1169–1175. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 2012, 1, e00205. [Google Scholar] [CrossRef]
- Lehmann, L.; Ferrari, R.; Vashisht, A.A.; Wohlschlegel, J.A.; Kurdistani, S.K.; Carey, M. Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J. Biol. Chem. 2012, 287, 35784–35794. [Google Scholar] [CrossRef] [Green Version]
- Tzatsos, A.; Paskaleva, P.; Lymperi, S.; Contino, G.; Stoykova, S.; Chen, Z.; Wong, K.K.; Bardeesy, N. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J. Biol. Chem. 2011, 286, 33061–33069. [Google Scholar] [CrossRef] [Green Version]
- Gulay, K.C.M.; Aoshima, K.; Shibata, Y.; Yasui, H.; Yan, Q.; Kobayashi, A.; Kimura, T. KDM2B promotes cell viability by enhancing DNA damage response in canine hemangiosarcoma. J. Genet. Genomics 2021, 48, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, X.; Wang, H.; Shao, Q. The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 2018, 10, 2222–2233. [Google Scholar]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Rakowsky, E.; Katz, A.; Gutman, H.; Sulkes, A.; Schacter, J.; Fenig, E. Tailoring treatment for classical Kaposi’s sarcoma: Comprehensive clinical guidelines. Int. J. Oncol. 1999, 14, 1097–1102. [Google Scholar] [CrossRef]
- Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Decouvelaere, A.V.; Péoc’h, M.; Istier, L.; Chalabreysse, P.; Muller, C.; Alberti, L.; Bringuier, P.P.; et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS ONE 2011, 6, e20294. [Google Scholar] [CrossRef]
- Si, H.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 2006, 80, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, F.Q.; Compitello, N.; Horwitz, E.; Sramkoski, M.; Knudsen, E.S.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J. Biol. Chem. 2005, 280, 3862–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [Green Version]
- Fujimuro, M.; Liu, J.; Zhu, J.; Yokosawa, H.; Hayward, S.D. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Virol. 2005, 79, 10429–10441. [Google Scholar] [CrossRef] [Green Version]
- Fujimuro, M.; Hayward, S.D. Manipulation of glycogen-synthase kinase-3 activity in KSHV-associated cancers. J. Mol. Med. 2004, 82, 223–231. [Google Scholar] [CrossRef]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, D.L.; Cannon, M.; Liu, Y.F.; Renne, R.; Chadburn, A.; Boshoff, C.; Cesarman, E. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 2008, 111, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Wada, S.; Eguchi, H.; Adachi, J.; Mishima, K.; Matsutani, M.; Nishikawa, R.; Nishiyama, M. Cadherin 13 overexpression as an important factor related to the absence of tumor fluorescence in 5-aminolevulinic acid-guided resection of glioma. J. Neurosurg. 2013, 119, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.V.; Kutuzov, M.A. Cadherin 13 in cancer. Genes Chromosomes Cancer 2010, 49, 775–790. [Google Scholar] [CrossRef]
- Li, W.; Wang, Q.; Qi, X.; Guo, Y.; Lu, H.; Chen, Y.; Lu, Z.; Yan, Q.; Zhu, X.; Jung, J.U.; et al. Viral interleukin-6 encoded by an oncogenic virus promotes angiogenesis and cellular transformation by enhancing STAT3-mediated epigenetic silencing of caveolin 1. Oncogene 2020, 39, 4603–4618. [Google Scholar] [CrossRef] [PubMed]
- Racadot, E.; Audhuy, B.; Duvernoy, H.; Thyss, A.; Lang, J.M.; Wijdenes, J.; Hervé, P. Clinical and immunological follow-up of patients with AIDS-associated Kaposi’s sarcoma treated with an anti-IL-6 monoclonal antibody. Cytokines Mol. Ther. 1995, 1, 133–138. [Google Scholar] [PubMed]
- He, M.; Zhang, W.; Bakken, T.; Schutten, M.; Toth, Z.; Jung, J.U.; Gill, P.; Cannon, M.; Gao, S.J. Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 2012, 72, 3582–3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, S.C.; Roenigk, R.K. Leiomyosarcoma of the skin. Treatment of 34 cases. Dermatol. Surg. 1996, 22, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, M.E.; Georgescu, C.V.; Radu, C.C.; Georgescu, C.C.; Stoica, L.E. Cutaneous leiomyosarcoma—Case report. J. Med. Life 2014, 7, 270–273. [Google Scholar] [PubMed]
- Vargas, A.C.; Gray, L.A.; White, C.L.; Maclean, F.M.; Grimison, P.; Ardakani, N.M.; Bonar, F.; Algar, E.M.; Cheah, A.L.; Russell, P.; et al. Genome wide methylation profiling of selected matched soft tissue sarcomas identifies methylation changes in metastatic and recurrent disease. Sci. Rep. 2021, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.M.; Sharma, A.; Ruzgar, N.M.; Deshpande, H.; Olino, K.; Khan, S.; Ahuja, N. Epigenetic signatures differentiate uterine and soft tissue leiomyosarcoma. Oncotarget 2021, 12, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Shridhar, K.; Walia, G.K.; Aggarwal, A.; Gulati, S.; Geetha, A.V.; Prabhakaran, D.; Dhillon, P.K.; Rajaraman, P. DNA methylation markers for oral pre-cancer progression: A critical review. Oral Oncol. 2016, 53, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, X.; Brownell, J.E. Biochemical perspectives on targeting KMT2 methyltransferases in cancer. Trends Pharmacol. Sci. 2021, 42, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Poreba, E.; Lesniewicz, K.; Durzynska, J. Aberrant Activity of Histone-Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. Int. J. Mol. Sci. 2020, 21, 24. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- De Carvalho Fischer, C.; Hu, Y.; Morreale, M.; Lin, W.Y.; Wali, A.; Thakar, M.; Karunasena, E.; Sen, R.; Cai, Y.; Murphy, L.; et al. Treatment with epigenetic agents profoundly inhibits tumor growth in leiomyosarcoma. Oncotarget 2018, 9, 19379–19395. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Dalla, E.; Franforte, E.; Paluvai, H.; Minisini, M.; Trevisanut, M.; Picco, R.; Brancolini, C. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020, 48, 646–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monga, V.; Swami, U.; Tanas, M.; Bossler, A.; Mott, S.L.; Smith, B.J.; Milhem, M. A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas. Cancers 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engström, K.; Bergh, P.; Gustafson, P.; Hultborn, R.; Johansson, H.; Löfvenberg, R.; Zaikova, O.; Trovik, C.; Wahlström, O.; Bauer, H.C. Liposarcoma: Outcome based on the Scandinavian Sarcoma Group register. Cancer 2008, 113, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Mashima, E.; Sawada, Y.; Nakamura, M. Recent Advancement in Atypical Lipomatous Tumor Research. Int. J. Mol. Sci. 2021, 22, 994. [Google Scholar] [CrossRef]
- Mashima, E.; Sawada, Y.; Saito-Sasaki, N.; Yamamoto, K.; Ohmori, S.; Omoto, D.; Yoshioka, H.; Yoshioka, M.; Okada, E.; Aoki, T.; et al. A Retrospective Study of Superficial Type Atypical Lipomatous Tumor. Front. Med. 2020, 7, 609515. [Google Scholar] [CrossRef]
- Kammerer-Jacquet, S.F.; Thierry, S.; Cabillic, F.; Lannes, M.; Burtin, F.; Henno, S.; Dugay, F.; Bouzillé, G.; Rioux-Leclercq, N.; Belaud-Rotureau, M.A.; et al. Differential diagnosis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma: Utility of p16 in combination with MDM2 and CDK4 immunohistochemistry. Hum. Pathol. 2017, 59, 34–40. [Google Scholar] [CrossRef]
- Knösel, T.; Altendorf-Hofmann, A.; Lindner, L.; Issels, R.; Hermeking, H.; Schuebbe, G.; Gibis, S.; Siemens, H.; Kampmann, E.; Kirchner, T. Loss of p16(INK4a) is associated with reduced patient survival in soft tissue tumours, and indicates a senescence barrier. J. Clin. Pathol. 2014, 67, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Manji, G.A.; Schwartz, G.K. Managing Liposarcomas: Cutting Through the Fat. J. Oncol. Pract. 2016, 12, 221–227. [Google Scholar] [CrossRef]
- Kanojia, D.; Nagata, Y.; Garg, M.; Lee, D.H.; Sato, A.; Yoshida, K.; Sato, Y.; Sanada, M.; Mayakonda, A.; Bartenhagen, C.; et al. Genomic landscape of liposarcoma. Oncotarget 2015, 6, 42429–42444. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, H.; Ishiguro, M.; Nishio, J.; Aoki, M.; Yokoyama, R.; Yokoyama, K.; Taguchi, K.; Nabeshima, K. Extensive lipoma-like changes of myxoid liposarcoma: Morphologic, immunohistochemical, and molecular cytogenetic analyses. Virchows Arch. 2015, 466, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, S.; Luo, P.; Yan, W.; Wang, C. Liposarcoma: Advances in Cellular and Molecular Genetics Alterations and Corresponding Clinical Treatment. J. Cancer 2020, 11, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, S.; Fritzsch, C.; Lehnhardt, M.; Zahn, S.; Kutzner, N.; Kuhnen, C.; Müller, O. Hypermethylation of the APC promoter but lack of APC mutations in myxoid/round-cell liposarcoma. Int. J. Cancer 2006, 119, 2347–2352. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Aisner, S.; Benevenia, J.; Patterson, F.; Harrison, L.E.; Hameed, M. Epigenetic alteration of p16INK4a gene in dedifferentiation of liposarcoma. Pathol. Res. Pract. 2009, 205, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; DeCarolis, P.L.; Angeles, C.V.; Brenet, F.; Schultz, N.; Antonescu, C.R.; Scandura, J.M.; Sander, C.; Viale, A.J.; Socci, N.D.; et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 2011, 1, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Seligson, N.D.; Stets, C.W.; Demoret, B.W.; Awasthi, A.; Grosenbacher, N.; Shakya, R.; Hays, J.L.; Chen, J.L. Inhibition of histone deacetylase 2 reduces MDM2 expression and reduces tumor growth in dedifferentiated liposarcoma. Oncotarget 2019, 10, 5671–5679. [Google Scholar] [CrossRef]
- Nakazawa, M.S.; Eisinger-Mathason, T.S.; Sadri, N.; Ochocki, J.D.; Gade, T.P.; Amin, R.K.; Simon, M.C. Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat. Commun. 2016, 7, 10539. [Google Scholar] [CrossRef] [Green Version]
- Keung, E.Z.; Akdemir, K.C.; Al Sannaa, G.A.; Garnett, J.; Lev, D.; Torres, K.E.; Lazar, A.J.; Rai, K.; Chin, L. Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. J. Clin. Investig. 2015, 125, 2965–2978. [Google Scholar] [CrossRef] [Green Version]
- Lazarides, A.L.; Eward, W.C.; Speicher, P.J.; Hou, C.H.; Nussbaum, D.P.; Green, C.; Blazer, D.G.; Kirsch, D.G.; Brigman, B.E. The Use of Radiation Therapy in Well-Differentiated Soft Tissue Sarcoma of the Extremities: An NCDB Review. Sarcoma 2015, 2015, 186581. [Google Scholar] [CrossRef] [Green Version]
- Antwih, D.A.; Gabbara, K.M.; Lancaster, W.D.; Ruden, D.M.; Zielske, S.P. Radiation-induced epigenetic DNA methylation modification of radiation-response pathways. Epigenetics 2013, 8, 839–848. [Google Scholar] [CrossRef]
- Herberg, M.; Siebert, S.; Quaas, M.; Thalheim, T.; Rother, K.; Hussong, M.; Altmüller, J.; Kerner, C.; Galle, J.; Schweiger, M.R.; et al. Loss of Msh2 and a single-radiation hit induce common, genome-wide, and persistent epigenetic changes in the intestine. Clin. Epigenetics 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.; Ando, K.; Nishikori, M.; Shibayama, H.; Teshima, T.; Kuroda, J.; Kato, K.; Imaizumi, Y.; Nosaka, K.; Sakai, R.; et al. Phase II study of tazemetostat for relapsed or refractory B-cell non-Hodgkin lymphoma with EZH2 mutation in Japan. Cancer Sci. 2021, 112, 3627–3635. [Google Scholar] [CrossRef]
- Munakata, W.; Shirasugi, Y.; Tobinai, K.; Onizuka, M.; Makita, S.; Suzuki, R.; Maruyama, D.; Kawai, H.; Izutsu, K.; Nakanishi, T.; et al. Phase 1 study of tazemetostat in Japanese patients with relapsed or refractory B-cell lymphoma. Cancer Sci. 2021, 112, 1123–1131. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Sarkozy, C.; Morschhauser, F.; Dubois, S.; Molina, T.; Michot, J.M.; Cullières-Dartigues, P.; Suttle, B.; Karlin, L.; Le Gouill, S.; Picquenot, J.M.; et al. A LYSA Phase Ib Study of Tazemetostat (EPZ-6438) plus R-CHOP in Patients with Newly Diagnosed Diffuse Large B-Cell Lymphoma (DLBCL) with Poor Prognosis Features. Clin. Cancer Res. 2020, 26, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Stephenson, J.; Hotte, S.; Nemunaitis, J.; Bélanger, K.; Reid, G.; Martell, R.E. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Investig. 2012, 30, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Saunthararajah, Y.; Sekeres, M.; Advani, A.; Mahfouz, R.; Durkin, L.; Radivoyevitch, T.; Englehaupt, R.; Juersivich, J.; Cooper, K.; Husseinzadeh, H.; et al. Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. J. Clin. Investig. 2015, 125, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, S.; Zhao, Y.; Kang, B.; Chen, C.; Chen, X.; Zhang, X.; Chen, W.; Dan, S.; Wang, H.; Wang, Y.J.; et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020, 11, 724. [Google Scholar] [CrossRef]
- Payet, M.; Dargai, F.; Gasque, P.; Guillot, X. Epigenetic Regulation (Including Micro-RNAs, DNA Methylation and Histone Modifications) of Rheumatoid Arthritis: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, X.; Li, H. Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 2018, 53, 169–177. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Jostes, S.; Nettersheim, D.; Schorle, H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat. Rev. Urol. 2019, 16, 245–259. [Google Scholar] [CrossRef]
- Wu, L.; Zeng, S.; Cao, Y.; Huang, Z.; Liu, S.; Peng, H.; Zhi, C.; Ma, S.; Hu, K.; Yuan, Z. Inhibition of HDAC4 Attenuated JNK/c-Jun-Dependent Neuronal Apoptosis and Early Brain Injury Following Subarachnoid Hemorrhage by Transcriptionally Suppressing MKK7. Front. Cell Neurosci. 2019, 13, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravillah, D.; Mohammed, A.; Qian, L.; Brewer, M.; Zhang, Y.; Biddick, L.; Steele, V.E.; Rao, C.V. Chemopreventive effects of an HDAC2-selective inhibitor on rat colon carcinogenesis and APCmin/+ mouse intestinal tumorigenesis. J. Pharmacol. Exp. Ther. 2014, 348, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Jiao, F.Z.; Wang, Y.; Zhang, H.Y.; Zhang, W.B.; Wang, L.W.; Gong, Z.J. Histone Deacetylase 2 Inhibitor CAY10683 Alleviates Lipopolysaccharide Induced Neuroinflammation Through Attenuating TLR4/NF-κB Signaling Pathway. Neurochem. Res. 2018, 43, 1161–1170. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Chen, Q.; Jiao, F.; Wang, L.; Gong, Z. HDAC2 inhibitor CAY10683 reduces intestinal epithelial cell apoptosis by inhibiting mitochondrial apoptosis pathway in acute liver failure. Histol. Histopathol. 2019, 34, 1173–1184. [Google Scholar]
- Lubieniecka, J.M.; de Bruijn, D.R.; Su, L.; van Dijk, A.H.; Subramanian, S.; van de Rijn, M.; Poulin, N.; van Kessel, A.G.; Nielsen, T.O. Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res. 2008, 68, 4303–4310. [Google Scholar] [CrossRef] [Green Version]
- Laporte, A.N.; Barrott, J.J.; Yao, R.J.; Poulin, N.M.; Brodin, B.A.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. HDAC and Proteasome Inhibitors Synergize to Activate Pro-Apoptotic Factors in Synovial Sarcoma. PLoS ONE 2017, 12, e0169407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, T.; Mayer-Steinacker, R.; Mayer, F.; Grünwald, V.; Schütte, J.; Hartmann, J.T.; Kasper, B.; Hüsing, J.; Hajda, J.; Ottawa, G.; et al. Vorinostat in refractory soft tissue sarcomas—Results of a multi-centre phase II trial of the German Soft Tissue Sarcoma and Bone Tumour Working Group (AIO). Eur. J. Cancer 2016, 64, 74–82. [Google Scholar] [CrossRef]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; Zhang, X.L.; Liu, Z.X.; Zhao, J.J.; Pan, Q.Z.; Wen, X.Z.; Xiao, W.; Xu, B.S.; Hong, D.C.; Guo, T.H.; et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J. Immunother. Cancer 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cutaneous Sarcomas | Epigenetic Modulator | Histone Modification | DNA Modification |
|---|---|---|---|
| DFSP | EZH2↑ [30] | ||
| Angiosarcoma | KDM2B↑ [53] | H3K27me3↓ [46] | |
| Kaposi’s sarcoma | DNMT1↑ [69] DNMT3A↑ [66] EZH2↑ [71] | Methylation TbetaRII↑ [64] | |
| Leiomyosarcoma | EZH2↑ [75]. KMT2A-AFDN↑ [75] BRD4↑ [75] HDAC4↑ [75] HDAC9↑ [81] DNMT↑ [80] | Methylation DAXX↑ [74] PTPRN2↑ [74] | |
| Liposarcoma | miR-486↑ [92] HDAC2↑ [96] | H3K9me3↑ [98] | Methylation p16INK4a↑ [94] APC↑ [92] C/EBPα↑ [95]. |
| Candidates | Action Mechanism |
|---|---|
| Tazemetostat | EZH2 inhibitor [102,103,104,105,106,107] |
| MG98 | DNMT1 inhibitor [109] |
| Decitabine | DNMT1 inhibitor [110] |
| SGI-1027 | DNMT1 inhibitor [111] |
| Quisinostat and romidepsin | Pan-HDAC inhibitor [121,122] |
| OSU-HDAC42 | HDAC2 inhibitor [118] |
| CAY10683 | HDAC2 inhibitor [120] |
| LMK235 | HDAC4 inhibitor [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashima, E.; Sawada, Y. Epigenetics of Cutaneous Sarcoma. Int. J. Mol. Sci. 2022, 23, 422. https://doi.org/10.3390/ijms23010422
Mashima E, Sawada Y. Epigenetics of Cutaneous Sarcoma. International Journal of Molecular Sciences. 2022; 23(1):422. https://doi.org/10.3390/ijms23010422
Chicago/Turabian StyleMashima, Emi, and Yu Sawada. 2022. "Epigenetics of Cutaneous Sarcoma" International Journal of Molecular Sciences 23, no. 1: 422. https://doi.org/10.3390/ijms23010422
APA StyleMashima, E., & Sawada, Y. (2022). Epigenetics of Cutaneous Sarcoma. International Journal of Molecular Sciences, 23(1), 422. https://doi.org/10.3390/ijms23010422