
Letrozole Accelerates Metabolic Remodeling through Activation of Glycolysis in Cardiomyocytes: A Role beyond Hormone Regulation


Abstract
:1. Introduction
2. Results
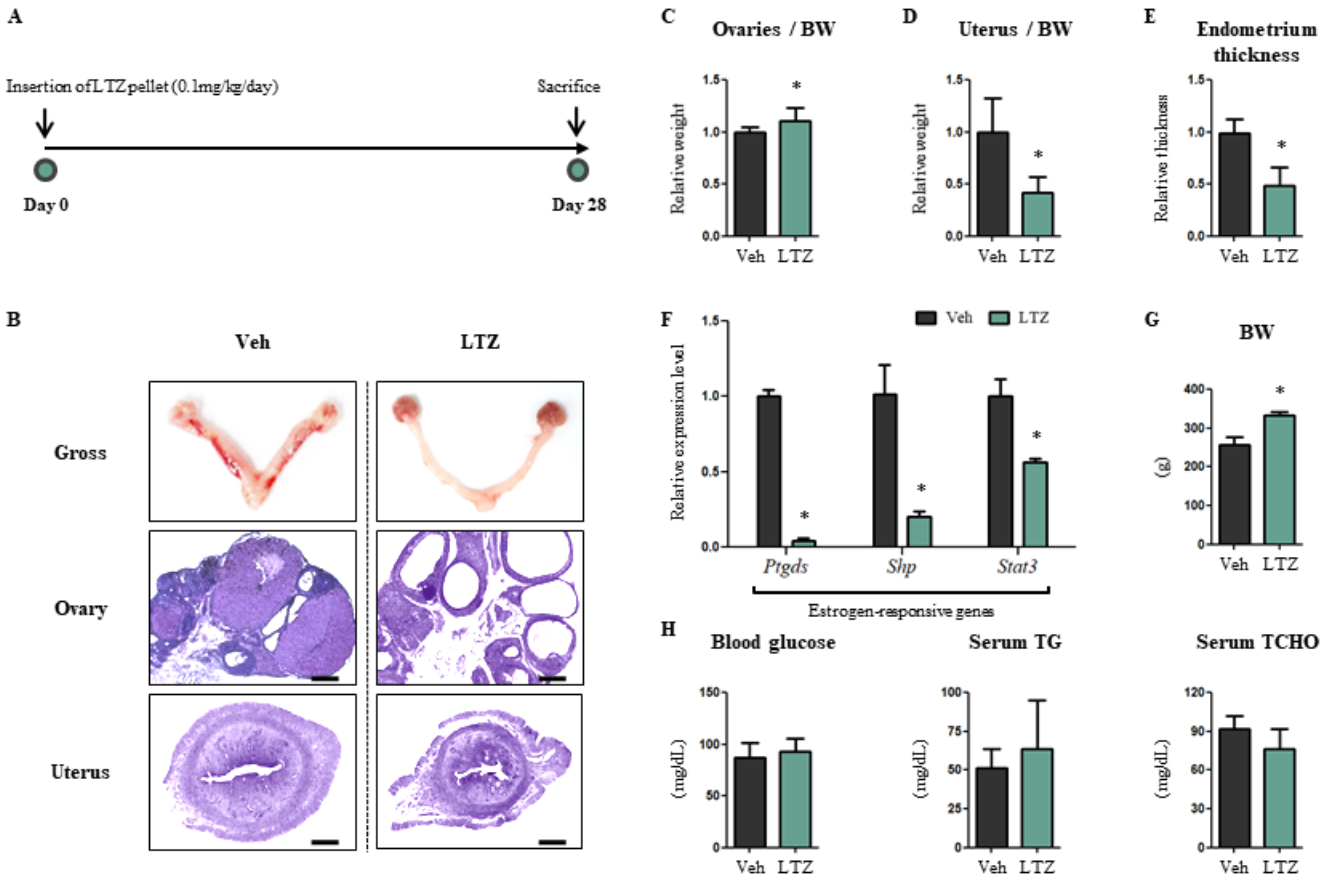
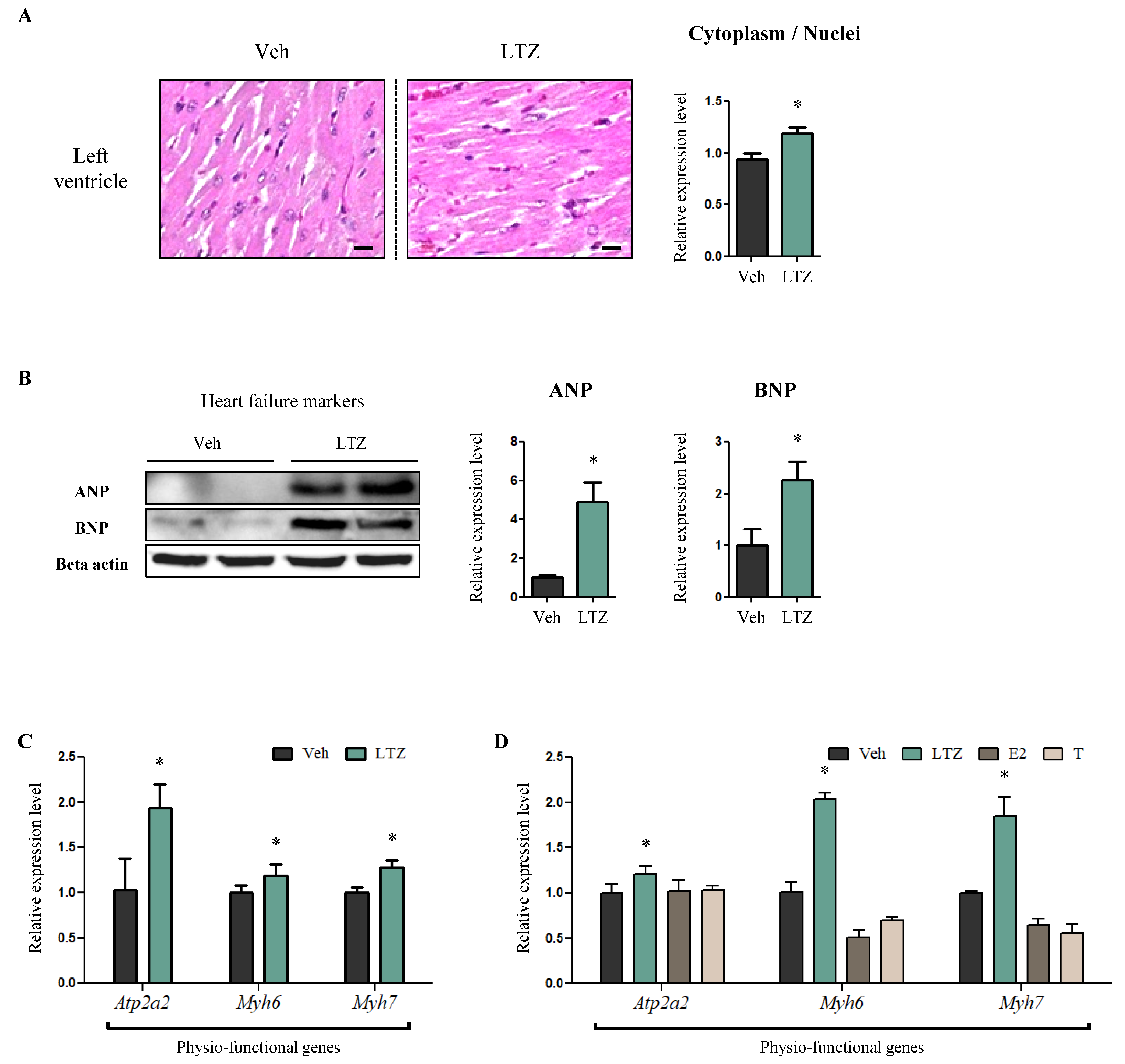
2.1. Letrozole Promotes Hypertrophic Cardiomyopathy of the Left Ventricle (LV) in Letrozole-Treated Rats (LTZ-Rats)
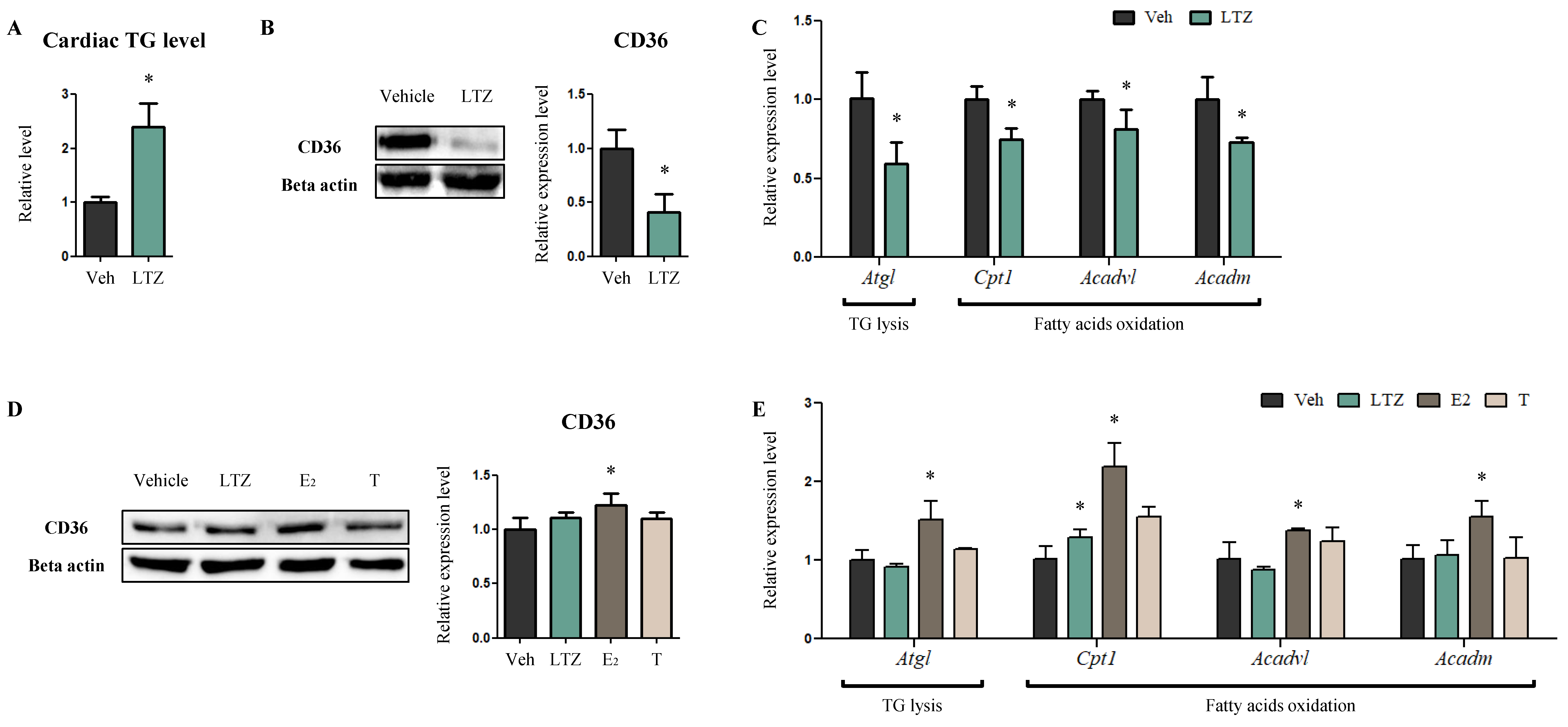
2.2. Letrozole Decreases FA Consumption in Cardiomyocytes While Regulating Estrogen Levels
2.3. Letrozole Induces Enhanced Glucose Consumption in Cardiomyocytes Independent of Its Role in Modulating Estrogen Levels
3. Discussion
4. Materials and Methods
4.1. Antibody
4.2. Animals
4.3. Hematological Measure Assay
4.4. Hematoxylin & Eosin Staining
4.5. Tissue Triglyceride Measure Assay
4.6. Cell Culture
4.7. Glucose Addition Assay
4.8. Glycolysis Stress Test
4.9. Total RNA Extraction and Real-Time Quantitative PCR
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mesa-Eguiagaray, I.; Wild, S.H.; Rosenberg, P.S.; Bird, S.M.; Brewster, D.H.; Hall, P.S.; Cameron, D.A.; Morrison, D.; Figueroa, J.D. Distinct temporal trends in breast cancer incidence from 1997 to 2016 by molecular subtypes: A population-based study of Scottish cancer registry data. Br. J. Cancer 2020, 123, 852–859. [Google Scholar] [CrossRef]
- Anderson, W.F.; Katki, H.A.; Rosenberg, P.S. Incidence of breast cancer in the United States: Current and future trends. J. Natl. Cancer Inst. 2011, 103, 1397–1402. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Griggs, J.J.; Prestrud, A.A.; Temin, S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J. Oncol. Pract. 2010, 6, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J.; Panel, M. Strategies for subtypes—Dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Lumachi, F.; Luisetto, G.; Basso, S.M.; Basso, U.; Brunello, A.; Camozzi, V. Endocrine therapy of breast cancer. Curr. Med. Chem. 2011, 18, 513–522. [Google Scholar] [CrossRef]
- Barnadas, A.; Estevez, L.G.; Lluch-Hernandez, A.; Rodriguez-Lescure, A.; Rodriguez-Sanchez, C.; Sanchez-Rovira, P. An overview of letrozole in postmenopausal women with hormone-responsive breast cancer. Adv. Ther. 2011, 28, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Aronow, W. Cardiotoxicity of cancer chemotherapy in clinical practice. Hosp. Pract. 2019, 47, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R., Jr.; das Dores Cruz, F.; Goncalves Brandao, S.M.; Rigaud, V.O.C.; Higuchi-Dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Lee, A.S.; Nagendran, J.; Xie, X.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Pretreatment with angiotensin-converting enzyme inhibitor improves doxorubicin-induced cardiomyopathy via preservation of mitochondrial function. J. Thorac. Cardiovasc. Surg. 2011, 142, 396–403.e393. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; O’Regan, R.M. Adjuvant Endocrine Therapy. Cancer Treat. Res. 2018, 173, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Khosrow-Khavar, F.; Filion, K.B.; Bouganim, N.; Suissa, S.; Azoulay, L. Aromatase Inhibitors and the Risk of Cardiovascular Outcomes in Women With Breast Cancer: A Population-Based Cohort Study. Circulation 2020, 141, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Qadir, H.; Amir, E.; Fischer, H.D.; Fu, L.; Austin, P.C.; Harvey, P.J.; Rochon, P.A.; Lee, D.S.; Anderson, G.M. The risk of myocardial infarction with aromatase inhibitors relative to tamoxifen in post-menopausal women with early stage breast cancer. Eur. J. Cancer 2016, 68, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Franchi, M.; Tritto, R.; Tarantini, L.; Navazio, A.; Corrao, G. Adjuvant Hormonotherapy and Cardiovascular Risk in Post-Menopausal Women with Breast Cancer: A Large Population-Based Cohort Study. Cancers 2021, 13, 2254. [Google Scholar] [CrossRef] [PubMed]
- Minta, W.; Palee, S.; Mantor, D.; Sutham, W.; Jaiwongkam, T.; Kerdphoo, S.; Pratchayasakul, W.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. Estrogen deprivation aggravates cardiometabolic dysfunction in obese-insulin resistant rats through the impairment of cardiac mitochondrial dynamics. Exp. Gerontol. 2018, 103, 107–114. [Google Scholar] [CrossRef]
- Apaijai, N.; Charoenphandhu, N.; Ittichaichareon, J.; Suntornsaratoon, P.; Krishnamra, N.; Aeimlapa, R.; Chattipakorn, S.C.; Chattipakorn, N. Estrogen deprivation aggravates cardiac hypertrophy in nonobese Type 2 diabetic Goto-Kakizaki (GK) rats. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Faulds, M.H.; Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.A. The diversity of sex steroid action: Regulation of metabolism by estrogen signaling. J. Endocrinol. 2012, 212, 3–12. [Google Scholar] [CrossRef]
- Szafran, H.; Smielak-Korombel, W. The role of estrogens in hormonal regulation of lipid metabolism in women. Przegl. Lek. 1998, 55, 266–270. [Google Scholar]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic Origins of Heart Failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. Fatty acid utilization in the hypertrophied and failing heart: Molecular regulatory mechanisms. Am. J. Med. Sci. 1999, 318, 36–42. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.G. A metabocentric view of cardiac remodeling. Curr. Opin. Physiol. 2019, 10, 43–48. [Google Scholar] [CrossRef]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomainen, T.; Tavi, P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp. Cell Res. 2017, 360, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Young, S.; Villet, O.; Shao, D.; Neto, F.C.; Bettcher, L.F.; Hsu, Y.A.; Kolwicz, S.C., Jr.; Raftery, D.; Tian, R. Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis. Circ. Res. 2020, 126, 182–196. [Google Scholar] [CrossRef]
- Yang, H.; Lee, S.Y.; Lee, S.R.; Pyun, B.J.; Kim, H.J.; Lee, Y.H.; Kwon, S.W.; Suh, D.H.; Lee, C.H.; Hong, E.J.; et al. Therapeutic Effect of Ecklonia cava Extract in Letrozole-Induced Polycystic Ovary Syndrome Rats. Front. Pharmacol. 2018, 9, 1325. [Google Scholar] [CrossRef]
- Lee, Y.H.; Yang, H.; Lee, S.R.; Kwon, S.W.; Hong, E.J.; Lee, H.W. Welsh Onion Root (Allium fistulosum) Restores Ovarian Functions from Letrozole Induced-Polycystic Ovary Syndrome. Nutrients 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lee, Y.H.; Lee, S.R.; Kaya, P.; Hong, E.J.; Lee, H.W. Traditional Medicine (Mahuang-Tang) Improves Ovarian Dysfunction and the Regulation of Steroidogenic Genes in Letrozole-Induced PCOS Rats. J. Ethnopharmacol. 2020, 248, 112300. [Google Scholar] [CrossRef]
- Basso, C.; Michaud, K.; d’Amati, G.; Banner, J.; Lucena, J.; Cunningham, K.; Leone, O.; Vink, A.; van der Wal, A.C.; Sheppard, M.N.; et al. Cardiac hypertrophy at autopsy. Virchows Arch. 2021, 479, 79–94. [Google Scholar] [CrossRef]
- Tan, V.P.; Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy 2015, 11, 963–964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. Correction to: PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakalov, V.; Reyes-Uribe, L.; Deshpande, R.; Maloy, A.L.; Shapiro, S.D.; Angus, D.C.; Chang, C.H.; Le Moyec, L.; Wendell, S.G.; Kaynar, A.M. Dichloroacetate-induced metabolic reprogramming improves lifespan in a Drosophila model of surviving sepsis. PLoS ONE 2020, 15, e0241122. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, G.; Mumusoglu, S.; Zengin, D.; Karabulut, E.; Yildiz, B.O. The prevalence and phenotypic features of polycystic ovary syndrome: A systematic review and meta-analysis. Hum. Reprod. 2016, 31, 2841–2855. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Sanchez, L.A.; Knochenhauer, E.S.; Moran, C.; Lazenby, J.; Stephens, K.C.; Taylor, K.; Boots, L.R. Androgen excess in women: Experience with over 1000 consecutive patients. J. Clin. Endocrinol. Metab. 2004, 89, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Heo, J.H.; Jo, S.L.; Kim, G.; Kim, S.J.; Yoo, H.J.; Lee, K.P.; Kwun, H.J.; Shin, H.J.; Baek, I.J.; et al. Progesterone receptor membrane component 1 reduces cardiac steatosis and lipotoxicity via activation of fatty acid oxidation and mitochondrial respiration. Sci. Rep. 2021, 11, 8781. [Google Scholar] [CrossRef]
- Harvey, P.A.; Leinwand, L.A. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J. Cell Biol. 2011, 194, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.G.; Bhanot, K.; Kothary, R.; Boudreau-Lariviere, C. Hearts of dystonia musculorum mice display normal morphological and histological features but show signs of cardiac stress. PLoS ONE 2010, 5, e9465. [Google Scholar] [CrossRef]
- Campbell, S.E.; Mehan, K.A.; Tunstall, R.J.; Febbraio, M.A.; Cameron-Smith, D. 17beta-estradiol upregulates the expression of peroxisome proliferator-activated receptor alpha and lipid oxidative genes in skeletal muscle. J. Mol. Endocrinol. 2003, 31, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Djouadi, F.; Brandt, J.M.; Weinheimer, C.J.; Leone, T.C.; Gonzalez, F.J.; Kelly, D.P. The role of the peroxisome proliferator-activated receptor alpha (PPAR alpha) in the control of cardiac lipid metabolism. Prostaglandins Leukot. Essent. Fatty Acids 1999, 60, 339–343. [Google Scholar] [CrossRef]
- Oka, S.; Zhai, P.; Yamamoto, T.; Ikeda, Y.; Byun, J.; Hsu, C.P.; Sadoshima, J. Peroxisome Proliferator Activated Receptor-alpha Association With Silent Information Regulator 1 Suppresses Cardiac Fatty Acid Metabolism in the Failing Heart. Circ. Heart Fail. 2015, 8, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.S.; Frank, A.P.; Fatima, L.A.; Palmer, B.F.; Oz, O.K.; Clegg, D.J. Activation of estrogen receptor alpha induces beiging of adipocytes. Mol. Metab. 2018, 18, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Foryst-Ludwig, A.; Haemmerle, G.; Zechner, R. The Role of Adipose Triglyceride Lipase and Cytosolic Lipolysis in Cardiac Function and Heart Failure. Cell Rep. Med. 2020, 1, 100001. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, E.; Bierla, J.B.; Toczek, M.; Tyrankiewicz, U.; Pajak, B.; Domal-Kwiatkowska, D.; Ratajska, A.; Smolenski, R.T.; Mende, U.; Chlopicki, S. Narrow time window of metabolic changes associated with transition to overt heart failure in Tgaq*44 mice. Pharmacol. Rep. 2016, 68, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, K.; Okada, T.; Miyaura, C.; Saibara, T. Fenofibrate, a ligand for PPARalpha, inhibits aromatase cytochrome P450 expression in the ovary of mouse. J. Lipid Res. 2003, 44, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Salama, S.A.; Mohammad, M.A.; Diaz-Arrastia, C.R.; Kamel, M.W.; Kilic, G.S.; Ndofor, B.T.; Abdel-Baki, M.S.; Theiler, S.K. Estradiol-17beta upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-alpha and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J. Clin. Endocrinol. Metab. 2014, 99, 3790–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, D.; Kulkarni, S. Butein induces intrinsic pathway of apoptosis, vimentin proteolysis, and inhibition of cancer stem cell population in a human papillary thyroid cancer cell line. Toxicol. In Vitro 2021, 77, 105244. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Type | Cat. | Inc. |
|---|---|---|---|
| Beta actin | Mouse monoclonal | sc-47778 | Santa Cruz biotechology |
| ANP | Rabbit polyclonal | ab14348 | Abcam PLC |
| BNP | Rabbit polyclonal | ab19645 | Abcam PLC |
| CD36 | Rabbit polyclonal | A5792 | Company ABclonal, Inc. |
| HK2 | Rabbit polyclonal | A0994 | Company ABclonal, Inc. |
| PKM2 | Rabbit monoclonal | #4053T | Cell signaling technology |
| PDH | Rabbit polyclonal | A17432 | Company ABclonal, Inc. |
| Secondary antibody | Type | Code. | Inc. |
| Anti-Mouse IgG | Goat | 115-035-174 | Jackonimmuno |
| Anti-Rabbit IgG | Mouse | 211-032-171 | Jackonimmuno |
| Gene Name | Upper Primer (5′-3′) | Lower Primer (5′-3′) | Species |
|---|---|---|---|
| Rplp0 | AAA GGG TCC TGG CTT TGT CT | CCG ACT CTT CCT TTG CTT CG | Rat |
| Ptgds | GGT TCC GGG AGA AGA AAG AG | CAC TGA GAG GGA GTG GAA GC | Rat |
| Shp | CCT TGG ATG TCC TAG GCA AG | CAC CAC TGT TGG GTT CCT CT | Rat |
| Stat3 | ATG AAG AGT GCC TTC GTG GT | TGT TCG TGC CCA GAA TGT TA | Rat |
| Atp2a2 | CTG TGG AAA CCC TTG GTT GT | CTC CAA TGG GTG CAT AGG TT | Rat |
| Myh6 | ATG ACC TTC AGG CTG AGG AA | CTC TCC TGG GTC AGC TTC AG | Rat |
| Myh7 | GGG TAT CCG CAT CTG TAG GA | TTG GTG TGG CCA AAC TTG TA | Rat |
| Atgl | TAC TGA AGA CCC TGC CTG CT | TGG CAA GTT GTC TGA AAT GC | Rat |
| Cpt1 | AGG CCT CCA TGA CAA GAA TG | GTC ATG GCT AGG CGG TAC AT | Rat |
| Acadvl | TGA CCC TGC CAA GAA TGA CT | GTC ATG CAT GCC CAC AAT CT | Rat |
| Acadm | CCG GGA CTA GGG TTT AGC TT | ACT CTC CGG AAT GTG TGT GT | Rat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.H.; Lee, S.R.; Jo, S.L.; Yang, H.; Lee, H.W.; Hong, E.-J. Letrozole Accelerates Metabolic Remodeling through Activation of Glycolysis in Cardiomyocytes: A Role beyond Hormone Regulation. Int. J. Mol. Sci. 2022, 23, 547. https://doi.org/10.3390/ijms23010547
Heo JH, Lee SR, Jo SL, Yang H, Lee HW, Hong E-J. Letrozole Accelerates Metabolic Remodeling through Activation of Glycolysis in Cardiomyocytes: A Role beyond Hormone Regulation. International Journal of Molecular Sciences. 2022; 23(1):547. https://doi.org/10.3390/ijms23010547
Chicago/Turabian StyleHeo, Jun H., Sang R. Lee, Seong Lae Jo, Hyun Yang, Hye Won Lee, and Eui-Ju Hong. 2022. "Letrozole Accelerates Metabolic Remodeling through Activation of Glycolysis in Cardiomyocytes: A Role beyond Hormone Regulation" International Journal of Molecular Sciences 23, no. 1: 547. https://doi.org/10.3390/ijms23010547
APA StyleHeo, J. H., Lee, S. R., Jo, S. L., Yang, H., Lee, H. W., & Hong, E. -J. (2022). Letrozole Accelerates Metabolic Remodeling through Activation of Glycolysis in Cardiomyocytes: A Role beyond Hormone Regulation. International Journal of Molecular Sciences, 23(1), 547. https://doi.org/10.3390/ijms23010547