
Enhancing the Therapeutic Potential of Mesenchymal Stromal Cell-Based Therapies with an Anti-Fibrotic Agent for the Treatment of Chronic Kidney Disease


Abstract
:1. Introduction
2. Chronic Kidney Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GFR and ACR Categories and Risk of Adverse Outcomes | Kidney Damage Stage:
ACR Categories (mg/g) | ||||
|---|---|---|---|---|---|
| <30 mg/g | 30–300 mg/g | >300 mg/g | |||
| A1 | A2 | A3 | |||
| Kidney Function Stage:GFR (mL/min/1.73m2) | ≥90 | Stage G1 | LR | MR | HR |
| 60–89 | Stage G2 | LR | MR | HR | |
| 45–59 | Stage G3a | MR | HR | VHR | |
| 30–44 | Stage G3b | HR | VHR | VHR | |
| 15–29 | Stage G4 | VHR | VHR | VHR | |
| <15 | Stage G5 | VHR | VHR | VHR | |
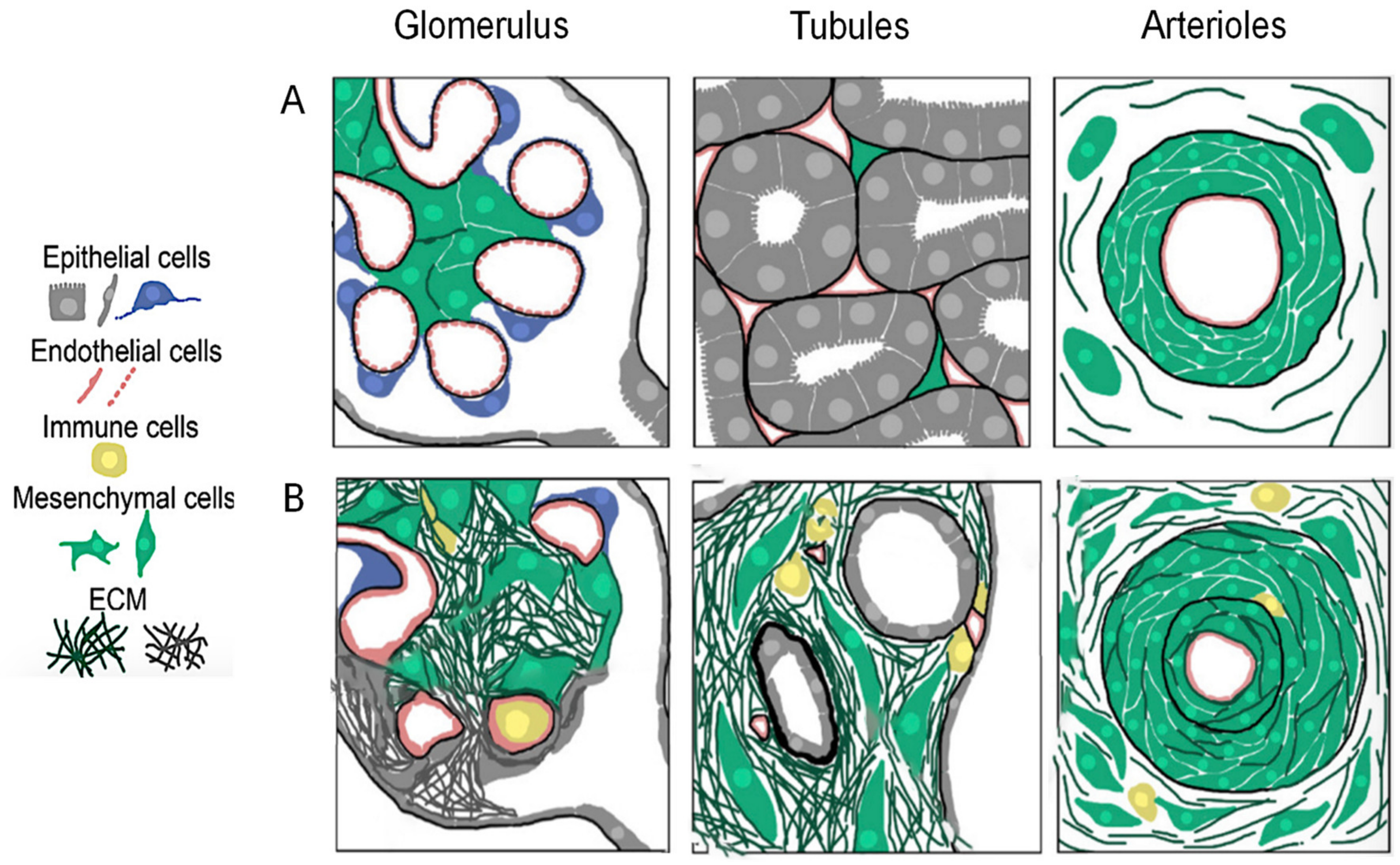
2.1. Pathological Events Underlying CKD
2.1.1. Hypertension
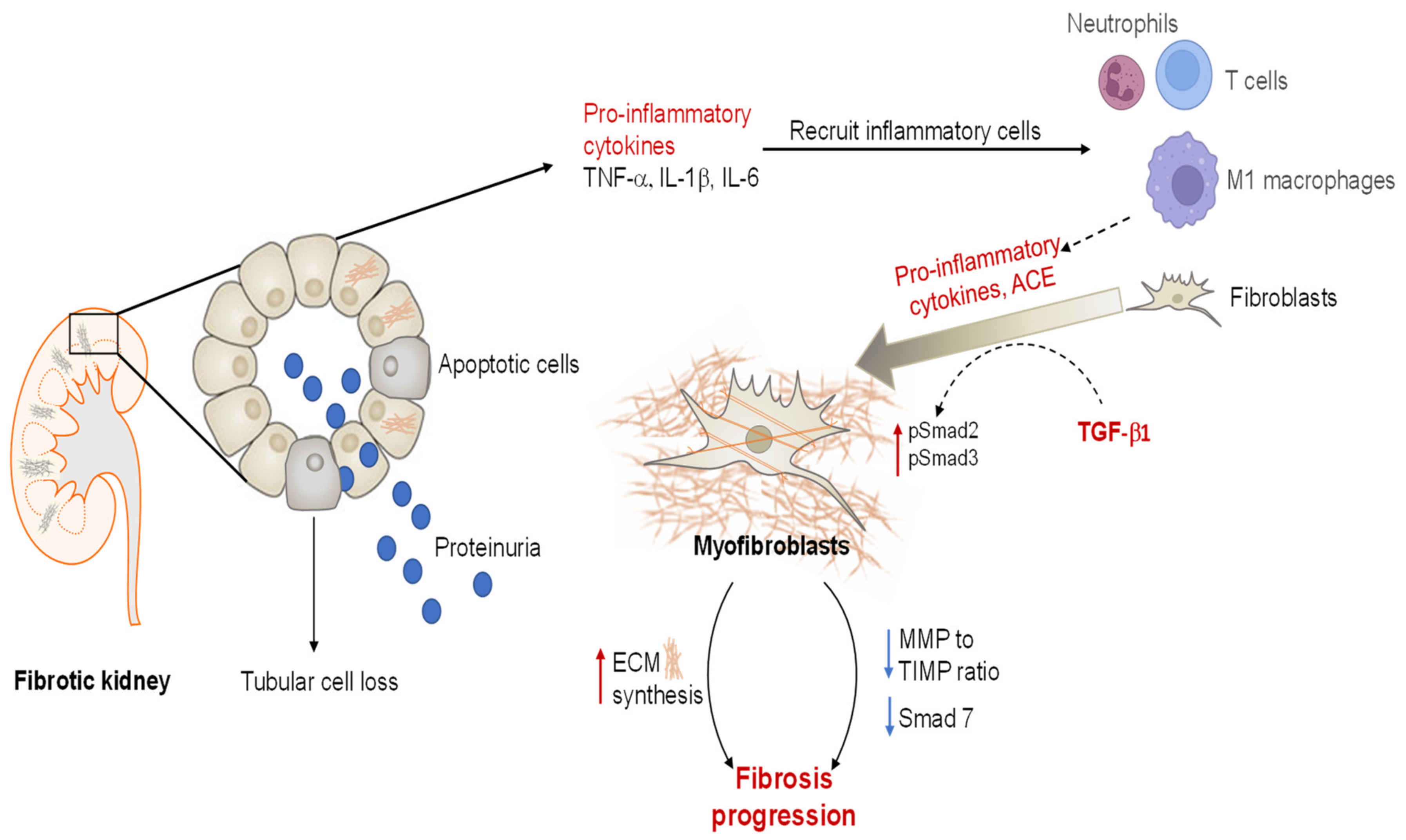
2.1.2. Renal Fibrosis
2.1.3. Angiotensin II and TGF-β1
3. Mesenchymal-Stem-Cell-Based Therapy
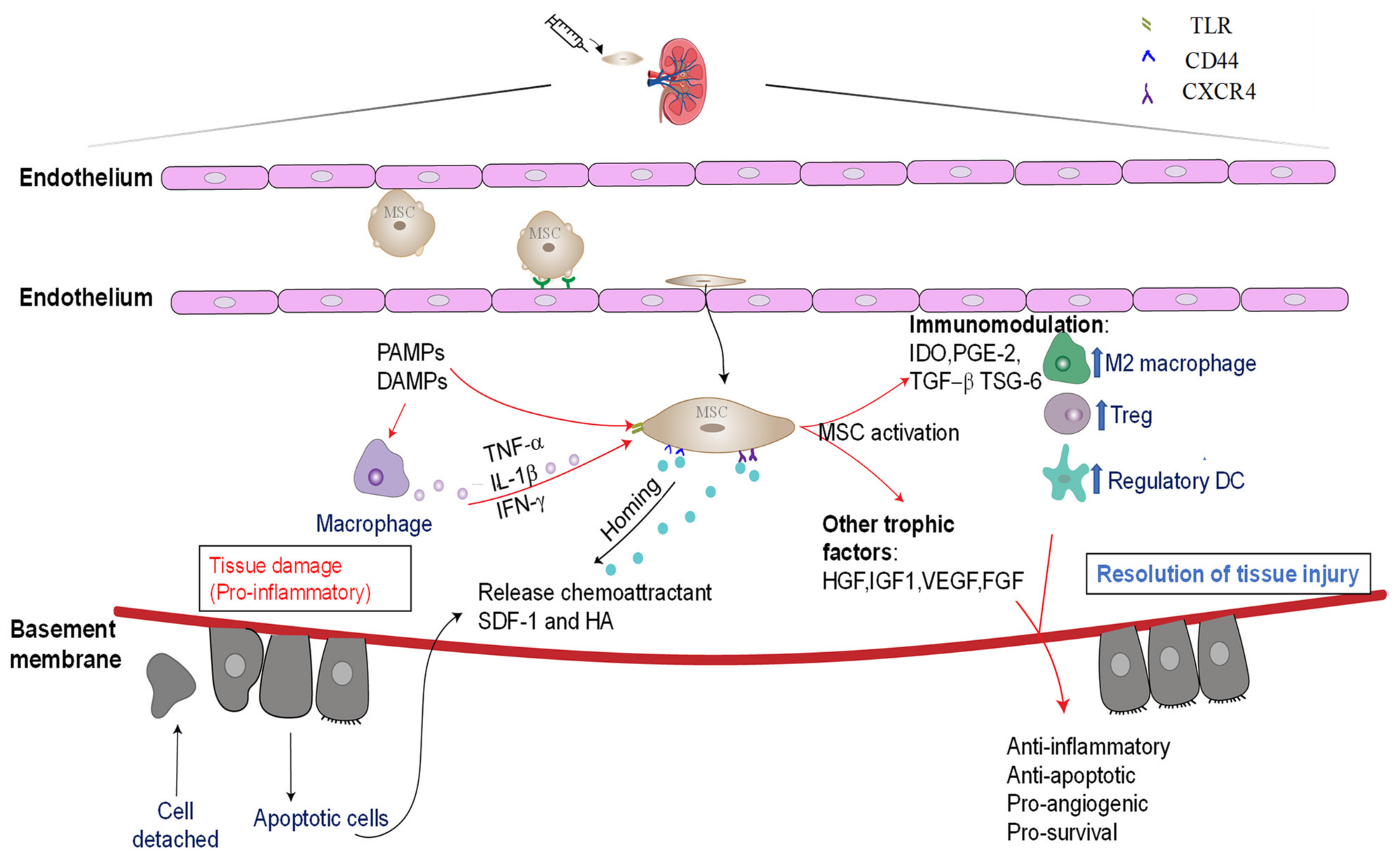
3.1. Mechanisms Underlying the Reparative Effects of BM-MCSs
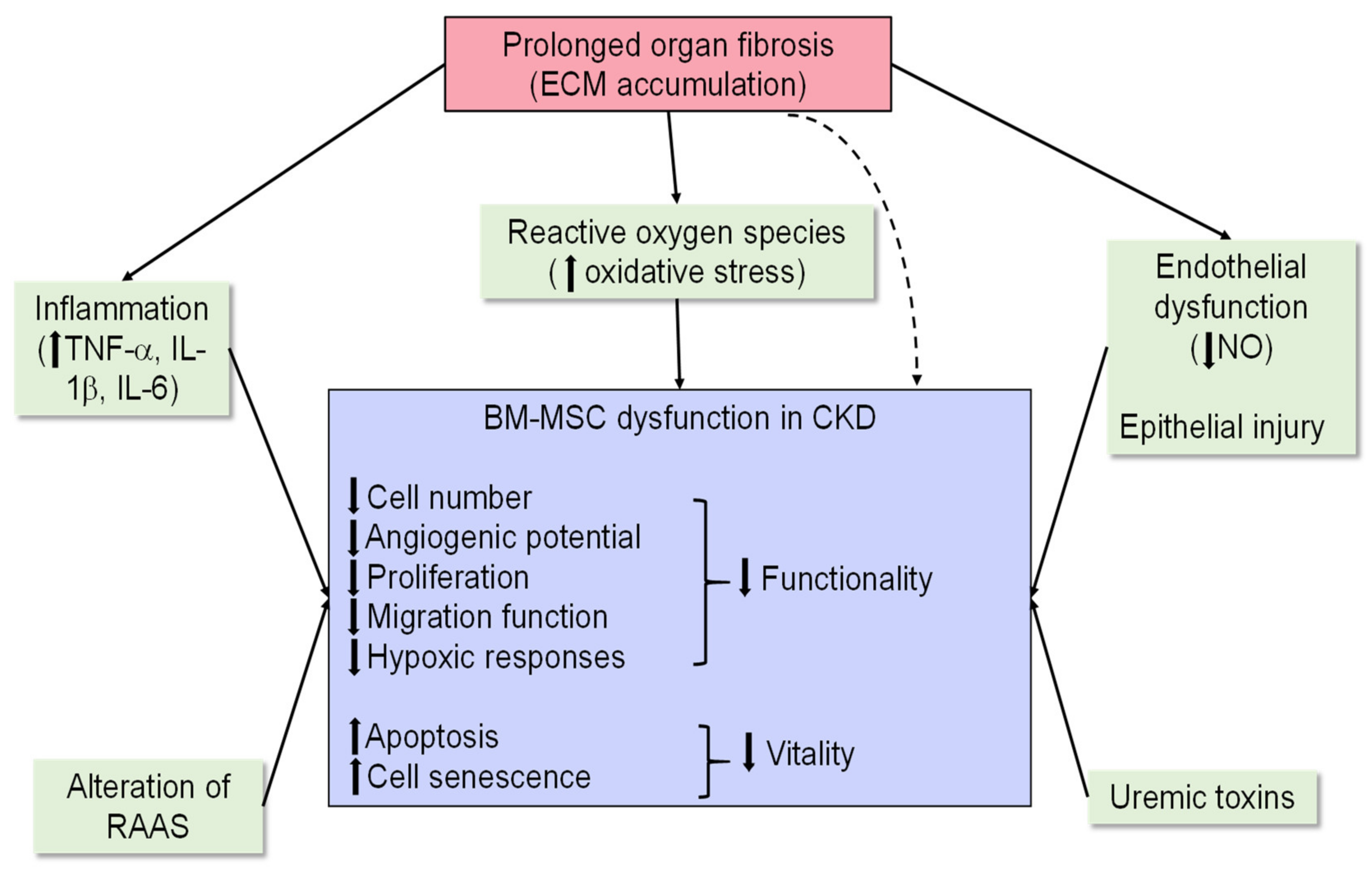
3.2. Preclinical Evidence for BM-MSCs as a Treatment Option for Chronic Models of Kidney Injury
| Etiology | In Vivo Models | MSC Number and Source | Routes of Delivery | Main Outcomes | Reference(s) |
|---|---|---|---|---|---|
| Hypertension | 2K1C induced renovascular hypertension | 1 × 106 rat BM-MSCs | Subcapsular injection | ↓ SBP ↓ Renin, ACE, and AT1R expression ↓ Renal Na+/K+ ATPase activity ↓ TGF-β1 and fibrosis ↓ Proteinuria ↑ AT2R expression ↑ Kidney morphology | [79] |
| 2K1C induced renovascular hypertension | 2 × 105 rat BM-MSCs | iv injection | ↓ SBP ↓ Sympathetic hyperactivity ↓ Angiotensinogen, ACE, and AT1R levels ↓ Fibrosis, inflammation, and proteinuria | [73] | |
| 2K1C induced renovascular hypertension | 1 × 106 rat BM-MSCs | iv injection | ↓ Inflammation and oxidative stress ↓ Morphological and ultrastructural abnormalities ↓ Serum urea and creatinine | [80] | |
| High-salt diet (8% NaCI) | 5 × 106 rat BM-MSCs | Intra-renal infusion | ↓ SBP ↓ Inflammasome activation ↓ Hypertensive kidney damage | [81] | |
| 5/6 subtotal nephrectomy | 2 × 105 rat BM-MSCs | iv injection | ↓ Fibrosis indices (collagen I, vimentin, TGF-β, α-SMA) ↓ Inflammation | [66] | |
| 5/6 subtotal nephrectomy | 2 × 105 rat BM-MSCs | Subcapsular injection | ↓ SBP ↑ Renal function (↓ Albuminuria, serum creatinine, GS) | [82] | |
| 1K/DOCA/salt | 1 × 106 human BM-MSCs | iv injection | ↓ SBP ↑ Renal function (↓ Proteinuria,↑creatinine clearance) and morphology ↓ Inflammation and fibrosis | [72,78] | |
| Obstructive nephropathy | Unilateral ureteric obstruction (UUO) | 1 × 106 human BM-MSCs | iv injection | ↓ Inflammation No anti-fibrotic effect | [71] |
| Nephrotoxicity | Cisplatin-induced chronic kidney damage | 3 × 106 rat BM-MSCs | iv injection | ↓ Creatinine and urea ↓ Inflammation and fibrosis ↑ Hepatocyte growth factor (protective for renal epithelial cells) | [65] |
3.3. BM-MSC Therapy in Clinical Trials for Managing CKD
| Clinical Trial Number/Reference | Center | Study Details | No. of Patients | Main Outcomes |
|---|---|---|---|---|
| NCT02195323 [83] | Royan Institute, Tehran, Iran | iv injection of 2 × 106/kg autologous BM-MSCs Phase 1 | 7 | Safety: No cell-related adverse events were reported during 18 months of follow-up Efficacy: No remarkable changes were observed in eGFR and serum creatinine in BM-MSC-treated patients compared to the baseline levels |
| NCT02166489 [84] | Royan Institute, Tehran, Iran | iv injection of autologous BM-MSCs, 2 × 106 cells/kg Phase 1 | 6 | Safety: No MSC-related adverse events during 12 months of follow-up |
| NCT01576328 [85] | Mesoblast, Ltd., Melbourne, Australia | iv injection of allogenic BM-MSCs, 0.3 × 106, 1 × 106, or 2 × 106 cells/kg Phase 1/2 | 61 | Safety: No cell-related adverse events over 12 weeks Efficacy: Clinical glycated haemoglobin target of <7% was achieved in 33.3% of participants in the 2 × 106/kg group, versus none in the placebo group |
| NCT01843387 [86] | Mesoblast, Ltd., Melbourne, Australia Monash University, Clayton, Australia Melbourne Renal Research Group, Melbourne, Australia | iv injection of allogenous BM-MSCs, at 150 × 106 or 300 × 106 cells/kg Phase 1/2 | 30 | Safety: No cell-related adverse events Efficacy: A trend towards stabilizing eGFR at week 12, which was maintained until 60 weeks of follow-up |
| NCT00698191 [88] | Nanjing Medical University, China | iv injection of allogeneic BM-MSCs, at 1 × 106/kg iv | 15 | Safety: No cell-related adverse events Efficacy: Amelioration of disease activity, with improvement in serologic markers and renal function |
4. Strategies Used to Enhance BM-MSC-Based Therapies
Combining BM-MSCs with the Anti-Fibrotic Agent Serelaxin
| Models | Treatment Regime | Main Outcomes | Reference(s) |
|---|---|---|---|
| In vitro | |||
| BM-MSCs | Treated with 1–100 ng/mL RLX for 24 h or 72 h | ↑ BM-MSC proliferation at 1 ng/mL after 72 h ↑ BM-MSC migration at 10 and 100 ng/mL after 24 h | [71] |
| Human EPCs isolated from the blood of stage V ESKD patients | Combination of 25% BM-MSC-derived conditioned medium (CM) + 10 ng/mL RLX | ↑ EPC proliferation and wound closure over 24 h ↑ EPC capillary tube formation over 4 h | [139] |
| In vivo | |||
| UUO-induced obstructive nephropathy (7 days) | RLX (0.5 mg/kg/day) via sc implanted osmotic minipumps + iv injection of BM-MSCs (1 × 106 per mouse); immediately after UUO | ↓ Tubular epithelial injury ↓ Macrophage infiltration ↓ Myofibroblast accumulation ↓ Collagen concentration ↑ MMP-2 activity | [71] |
| 1K/DOCA/Salt-induced hypertension (21 days) | RLX (0.5 mg/kg/day) via sc implanted osmotic minipumps + iv injection of BM-MSCs (1 × 106 per mouse); on day 14 | ↓ SBP ↓ Tubular epithelial injury ↓ Inflammation, fibrosis, and proteinuria ↑ Creatinine clearance ↑ Peritubular capillary density * Provided broader reno-protection than RLX and/ or BM-MSC-derived EXO, perindopril, or spironolactone | [72,78] |
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breyer, M.D.; Susztak, K. Developing treatments for chronic kidney disease in the 21st century. Semin. Nephrol. 2016, 36, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, A.F.; Ricardo, S.D. Mesenchymal stem cells in kidney inflammation and repair. Nephrology 2012, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Yu, M.R.; Kim, H.J.; Jeon, J.S.; Kwon, S.H.; Jin, S.Y.; Lee, J.; Jang, J.; Park, J.O.; Ziyadeh, F.; et al. Uremia induces functional incompetence of bone marrow-derived stromal cells. Nephrol. Dial. Transpl. 2012, 27, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Eirin, A.; Lerman, L.O. Challenges and opportunities for stem cell therapy in patients with chronic kidney disease. Kidney Int. 2016, 89, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Hodgkinson, C.P.; Gomez, J.A.; Mirotsou, M.; Dzau, V.J. Genetic engineering of mesenchymal stem cells and its application in human disease therapy. Hum. Gene Ther. 2010, 21, 1513–1526. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; De Zeeuw, D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [Green Version]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global prevalence of chronic kidney disease—A systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfre (AIHW). Chronic Kidney Disease Prevalence among Australian Adults over Time. 2018. Available online: https://www.aihw.gov.au/reports/chronic-kidney-disease/chronic-kidney-disease-prevalence-adults/summary (accessed on 5 October 2021).
- Ahmed, A.; Campbell, R.C. Epidemiology of chronic kidney disease in heart failure. Heart Fail. Clin. 2008, 4, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decreased, G. Definition and classification of CKD. Kidney Int. 2013, 3, 19–62. [Google Scholar]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of renal fibrosis—Turning challenges into opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Nami, R.; Bruno, R.M.; Quatrini, I.; Nuti, R. Hypertension, left ventricular hypertrophy and chronic kidney disease. Heart Fail. Rev. 2011, 16, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udani, S.; Lazich, I.; Bakris, G.L. Epidemiology of hypertensive kidney disease. Nat. Rev. Nephrol. 2010, 7, 11–21. [Google Scholar] [CrossRef]
- Griffin, K.A.; Bidani, A.K. Pathophysiology of hypertensive renal damage. Hypertension 2004, 44, 595–601. [Google Scholar]
- Fogo, A.B. Mechanisms of progression of chronic kidney disease. Pediatr. Nephrol. 2007, 22, 2011–2022. [Google Scholar] [CrossRef] [Green Version]
- Bidani, A.K.; Griffin, K.A. Basic science: Hypertensive target organ damage. J. Am. Soc. Hypertens. 2015, 9, 235–257, quiz 8. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.S.; Summers, R.J.; Hewitson, T.D. Antifibrotic actions of serelaxin–new roles for an old player. Trends Pharmacol. Sci. 2016, 37, 485–497. [Google Scholar] [CrossRef]
- Urushihara, M.; Kobori, H. Intrarenal renin-angiotensin system activation in end-stage renal disease. Hypertens. Res. 2017, 40, 351–352. [Google Scholar] [CrossRef]
- Munoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; Gonzalez-Gomez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end–organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruster, C.; Wolf, G. Renin–angiotensin–aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II—induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Schorb, W.; Booz, G.W.; Dostal, D.E.; Conrad, K.M.; Chang, K.C.; Baker, K.M. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ. Res. 1993, 72, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Carthy, J.M. TGFβ signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, T.D.; Mookerjee, I.; Masterson, R.; Zhao, C.; Tregear, G.W.; Becker, G.J.; Samuel, C.S. Endogenous relaxin is a naturally occurring modulator of experimental renal tubulointerstitial fibrosis. Endocrinology 2007, 148, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.-H.; Remuzzi, G.; Snappin, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.F.E.; Schmieder, R.E.; McQueen, M.; Dyal, L.; Schumacher, H.; Pogue, J.; Wang, X.; Maggioni, A.; Budaj, A.; Chaithirapham, S.; et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): A multicentre, randomised, double-blind, controlled trial. Lancet 2008, 372, 547–553. [Google Scholar] [CrossRef]
- Pugh, D.; Gallacher, P.J.; Dhaun, N. Management of hypertension in chronic kidney disease. Drugs 2019, 79, 365–379. [Google Scholar]
- Roufosse, C.; Cook, H.T. Stem cells and renal regeneration. Nephron Exp. Nephrol. 2008, 109, e39–e45. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Tetta, C.; Camussi, G. Contribution of stem cells to kidney repair. Am. J. Nephrol. 2008, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventure, J.P. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 2003, 14 (Suppl. S1), S55–S61. [Google Scholar] [CrossRef] [Green Version]
- Garbern, J.C.; Lee, R.T. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell 2013, 12, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Ajay, A.K.; Zhao, L.; Vig, S.; Fujiwara, M.; Thakurela, S.; Jadhav, S.; Cho, A.; Chiu, I.J.; Ding, Y.; Ramachandran, K.; et al. Deletion of STAT3 from Foxd1 cell population protects mice from kidney fibrosis by inhibiting pericytes trans–differentiation and migration. Cell Rep. 2022, 38, 110473. [Google Scholar] [CrossRef]
- Perico, N.; Casiraghi, F.; Remuzzi, G. Clinical translation of mesenchymal stromal cell therapies in nephrology. J. Am. Soc. Nephrol. 2018, 29, 362–375. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Ozaki, K.; Sato, K.; Oh, I.; Meguro, A.; Tatara, R.; Muroi, K.; Ozawa, K. Mechanisms of Immunomodulation by Mesenchymal Stem Cells. Int. J. Hematol. 2007, 86, 5–7. [Google Scholar] [CrossRef]
- Yamaoka, T.; Mahara, A. Cell rolling column in purification and differentiation analysis of stem cells. Reactive Funct. Polymers 2011, 71, 362–366. [Google Scholar]
- Yun, C.W.; Lee, S.H. Potential and therapeutic efficacy of cell-based therapy using mesenchymal stem cells for acute/chronic kidney disease. Int. J. Mol. Sci. 2019, 20, 1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, N.L.; Sun, G.; McDonald, C.; Layton, D.; Moussa, L.; Emerson-Webber, A.; Veron, N.; Siatkis, C.; Herszfeld, D.; Price, J.; et al. Distinct immunomodulatory and migratory mechanisms underpin the therapeutic potential of human mesenchymal stem cells in autoimmune demyelination. Cell Transplant. 2013, 22, 1409–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honczarenko, M.; Le, Y.; Swierkowski, M.; Ghiran, I.; Glodek, A.M.; Silberstein, L.E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 2006, 24, 1034–1041. [Google Scholar] [CrossRef]
- Zhu, H.; Mitsuhashi, N.; Klein, A.; Barsky, L.W.; Weinberg, K.; Barr, M.L.; Demetriou, A.; Wu, G.D. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem Cells 2006, 24, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, S.; Fierabracci, A. Insights into the secretome of mesenchymal stem cells and its potential applications. Int. J. Mol. Sci. 2019, 20, 4597. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Benigni, A. Mesenchymal stem cells and kidney repair. Nephrol. Dial. Transplant. 2013, 28, 788–793. [Google Scholar] [CrossRef] [Green Version]
- Morigi, M.; Rota, C.; Remuzzi, G. Mesenchymal stem cells in kidney repair. In Mesenchymal Stem Cells: Methods and Protocols; Gnecchi, M., Ed.; Springer: New York, NY, USA, 2016; pp. 89–107. [Google Scholar]
- Watt, S.M.; Gullo, F.; van der Garde, M.; Markeson, D.; Camicia, R.; Khoo, C.P.; Zwaginga, J.J. The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br. Med. Bull. 2013, 108, 25–53. [Google Scholar] [CrossRef]
- Wu, H.J.; Yiu, H.Y.; Li, R.X.; Wong, D.W.L.; Leung, J.C.K.; Chan, L.Y.Y.; Zhang, Y.; Lian, Q.; Lin, M.; Tse, H.F.; et al. Mesenchymal stem cells modulate albumin–induced renal tubular inflammation and fibrosis. PLoS ONE 2014, 9, e90883. [Google Scholar] [CrossRef] [Green Version]
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013, 91, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, S.; Morbelli, S.; Morando, S.; Massollo, M.; Marini, C.; Bertoni, A.; Frassoni, F.; Bartolomé, S.T.; Sambuceti, G.; Traggiai, E.; et al. Mesenchymal stem cells impair in vivo T-cell priming by dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17384–17389. [Google Scholar] [CrossRef] [Green Version]
- English, K.; Barry, F.P.; Mahon, B.P. Murine mesenchymal stem cells suppress dendritic cell migration, maturation and antigen presentation. Immunol. Lett. 2008, 115, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenert, P. Targeting Toll–like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res. Therap. 2006, 8, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, L.; Karimi, M.H.; Kamali-Sarvestani, E.; Azarpira, N.; Shariati, M. The immunomodulatory effect of bone-marrow mesenchymal stem cells on expression of TLR3 and TLR9 in mice dendritic cells. Int. J. Organ. Transplant. Med. 2017, 8, 35–42. [Google Scholar] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Kim, J.; Hematti, P. Mesenchymal stem cell–educated macrophages: A novel type of alternatively activated macrophages. Exp. Hematol. 2009, 37, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Lee, R.H.; Bazhanov, N.; Oh, J.Y.; Prockop, D.J. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan–induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood 2011, 118, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Németh, K.; Leelahavanichkul, A.; Yuen, P.S.T.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Kota, D.J.; Wiggins, L.L.; Yoon, N.; Le, R.H. TSG-6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes 2013, 62, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.Y.; Lee, R.H.; Yu, J.M.; Ko, J.H.; Lee, H.J.; Ko, A.Y.; Roddy, G.W.; Prockop, D.J. Intravenous mesenchymal stem cells prevented rejection of allogeneic corneal transplants by aborting the early inflammatory response. Mol. Ther. 2012, 20, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, A.; Sturgeon, C.; Siatskas, M.; Ferrer, K.; McIntosh, K.; Patil, S.; Hardy, W.; Devine, S.; Ucker, D.; Deans, R.; et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 2002, 30, 42–48. [Google Scholar] [CrossRef]
- Elseweidy, M.M.; Askar, M.E.; Elswefy, S.E.; Shawky, M. Nephrotoxicity induced by cisplatin intake in experimental rats and therapeutic approach of using mesenchymal stem cells and spironolactone. Appl. Biochem. Biotech. 2018, 184, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Semedo, P.; Correa–Costa, M.; Cenedeze, M.A.; Costa Malheiros, D.M.A.; dos Reis, M.A.; Shimizu, M.H.; Seguro, A.C.; Pacheco-Silva, A.; Camara, N.O.S. Mesenchymal stem cells attenuate renal fibrosis through immune modulation and remodelling properties in a rat remnant kidney model. Stem Cells 2009, 27, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.-M.; Deng, X.-D.; Li, J.-D.; Li, D.-H.; Cao, H.; Liu, Z.-X.; Zhang, J. Arterially transplanted mesenchymal stem cells in a mouse reversible unilateral ureteral obstruction model: In vivo bioluminescence imaging and effects on renal fibrosis. Chin. Med. J. 2013, 126, 1890–1894. [Google Scholar]
- Franquesa, M.; Herrero, E.; Torras, J.; Ripoll, E.; Flaquer, M.; Gomà, M.; Lloberas, N.; Anegon, I.; Cruzado, J.M.; Grinyó, J.M.; et al. Mesenchymal stem cell therapy prevents interstitial fibrosis and tubular atrophy in a rat kidney allograft model. Stem Cells Dev. 2012, 21, 3125–3135. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef]
- Mias, C.; Lairez, O.; Trouche, E.; Roncalli, J.; Calise, D.; Seguelas, M.H.; Orderner, C.; Piercecchi–Marti, M.-D.; Auge, N.; Salvayre, A.N.; et al. Mesenchymal stem cells promote matrix metalloproteinase secretion by cardiac fibroblasts and reduce cardiac ventricular fibrosis after myocardial infarction. Stem Cells 2009, 27, 2734–2743. [Google Scholar] [CrossRef]
- Huuskes, B.M.; Wise, A.F.; Cox, A.J.; Lim, E.X.; Payne, N.L.; Kelly, D.J.; Samuel, C.S.; Ricardo, S.D. Combination therapy of mesenchymal stem cells and serelaxin effectively attenuates renal fibrosis in obstructive nephropathy. FASEB J. 2015, 29, 540–553. [Google Scholar] [CrossRef]
- Li, Y.; Chakraborty, A.; Broughton, B.R.S.; Ferens, D.; Widdop, R.E.; Ricardo, S.D.; Samuel, C.S. Comparing the renoprotective effects of BM-MSCs versus BM-MSC-exosomes, when combined with an anti-fibrotic drug, in hypertensive mice. Biomed. Pharmacother. 2021, 144, 112256. [Google Scholar] [CrossRef]
- Oliveira-Sales, E.B.; Maquigussa, E.; Semedo, P.; Pereira, L.G.; Ferreira, V.M.; Camara, N.O.; Bergamaschi, C.T.; Campos, R.R.; Boim, M.A. Mesenchymal stem cells (MSC) prevented the progression of renovascular hypertension, improved renal function and architecture. PLoS ONE 2013, 8, e78464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira-Sales, E.B.; Varela, V.A.; Maquigussa, E.; Borges, F.T.; Shimoura, C.G.; Gomes, G.; Campos, R.R.; Boim, M.A. Renovascular hypertension: Effects of mesenchymal stem cells in the contralateral hypertensive kidney in rats. Clin. Exp. Hypertens. 2016, 38, 586–593. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira-Sales, E.B.; Nishi, E.E.; Boim, M.A.; Dolnikoff, M.S.; Bergamaschi, C.T.; Campos, R.R. Upregulation of AT1R and iNOS in the rostral ventrolateral medulla (RVLM) is essential for the sympathetic hyperactivity and hypertension in the 2K-1C Wistar rat model. Am. J. Hypertens. 2010, 23, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Sales, E.B.; Boim, M.A. Mesenchymal stem cells and chronic renal artery stenosis. Am. J. Physiol. Renal Physiol. 2015, 310, F6–F9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinar, A.A.; Scott, T.E.; Huuskes, B.M.; Tapia Caceres, F.E.; Kemp–Harper, B.K.; Samuel, C.S. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol. Ther. 2020, 209, 107511. [Google Scholar] [CrossRef]
- Li, Y.; Shen, M.; Ferens, D.; Broughton, B.R.S.; Murthi, P.; Saini, S.; Widdop, R.E.; Ricardo, S.D.; Pinar, A.A.; Samuel, C.S. Combining mesenchymal stem cells with serelaxin provides enhanced renoprotection against 1K/DOCA/salt–induced hypertension. Br. J. Pharmacol. 2021, 178, 1164–1181. [Google Scholar] [CrossRef]
- Lira, R.; Oliveira, M.; Martins, M.; Silva, C.; Carvalho, S.; Stumbo, A.C.; Cortez, E.; Verdoorn, K.; Einicker–Lamas, M.; Thole, A.; et al. Transplantation of bone marrow–derived MSCs improves renal function and Na++K+-ATPase activity in rats with renovascular hypertension. Cell Tissue Res. 2017, 369, 287–301. [Google Scholar] [CrossRef]
- Mohamed, E.M.; Samak, M.A. Therapeutic potentials of mesenchymal stem cells on the renal cortex of experimentally induced hypertensive albino rats: Relevant role of Nrf2. Tissue Cell 2017, 49, 358–367. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, X.-X.; Wang, W.; Hu, J.; Li, P.-L.; Conley, S.; Li, N. Mesenchymal stem cell transplantation inhibited high salt–induced activation of the NLRP3 inflammasome in the renal medulla in Dahl S rats. Am. J. Physiol. 2016, 310, F621–F627. [Google Scholar] [CrossRef] [Green Version]
- Cavaglieri, R.C.; Martini, D.; Sogayar, M.C.; Noronha, I.L. Mesenchymal stem cells delivered at the subcapsule of the kidney ameliorate renal disease in the rat remnant kidney model. Transpl. Proc. 2009, 41, 947–951. [Google Scholar] [CrossRef]
- Makhlough, A.; Shekarchian, S.; Moghadasali, R.; Einollahi, B.; Dastgheib, M.; Janbabaee, G.; Hosseini, S.E.; Falah, N.; Abbasi, F.; Baharvand, H.; et al. Bone marrow-mesenchymal stromal cell infusion in patients with chronic kidney disease: A safety study with 18 months of follow-up. Cytotherapy 2018, 20, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Makhlough, A.; Shekarchian, S.; Moghadasali, R.; Einollahi, B.; Hosseini, S.E.; Jaroughi, N.; Bolurieh, T.; Baharvand, H.; Aghdami, N. Safety and tolerability of autologous bone marrow mesenchymal stromal cells in ADPKD patients. Stem Cell Res. Therap. 2017, 8, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skyler, J.S.; Fonseca, V.A.; Segal, K.R.; Rosenstock, J. Allogeneic mesenchymal precursor cells in type 2 diabetes: A randomized, placebo-controlled, dose-escalation safety and tolerability pilot study. Diabetes Care 2015, 38, 1742–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic mesenchymal precursor cells (MPC) in diabetic nephropathy: A randomized, placebo-controlled, dose escalation study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Ortega, L.; Schultz, D.; Lenz, O.; Pardo, V.; Contreras, G. Lupus nephritis: Pathologic features, epidemiology and a guide to therapeutic decisions. Lupus 2010, 19, 557–574. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, H.; Hua, B.; Wang, H.; Lu, L.; Shi, S.; Hou, Y.; Zeng, X.; Gilkeson, G.S.; Sin, L. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: A pilot clinical study. Ann. Rheum. Dis. 2010, 69, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Lalu, M.M.; McIntyre, L.; Pugliese, C.; Fergusson, D.; Winston, B.W.; Marshall, J.C.; Granton, J.; Stewart, D.J.; Canadian Critical Care Trials Group. Safety of cell therapy with mesenchymal stromal cells (safecell): A systematic review and meta-analysis of clinical trials. PLoS ONE 2012, 7, e47559. [Google Scholar] [CrossRef]
- Crop, M.J.; Korevaar, S.S.; De Kuiper, R.; Ijzermans, J.N.; Van Besouw, N.M.; Baan, C.C.; Weimar, W.; Hoogdujin, M.J. Human mesenchymal stem cells are susceptible to lysis by CD8+ T cells and NK cells. Cell Transplant. 2011, 20, 1547–1559. [Google Scholar] [CrossRef] [Green Version]
- Kunter, U.; Rong, S.; Boor, P.; Eitner, F.; Müller-Newen, G.; Djuric, Z.; van Roeyen, C.R.; Konieczny, A.; Ostendorf, T.; Villa, L.; et al. Mesenchymal stem cells prevent progressive experimental renal failure but maldifferentiate into glomerular adipocytes. J. Am. Soc. Nephrol. 2007, 18, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Prigozhina, T.B.; Khitrin, S.; Elkin, G.; Eizik, O.; Morecki, S.; Slavin, S. Mesenchymal stromal cells lose their immunosuppressive potential after allotransplantation. Exp. Hematol. 2008, 36, 1370–1376. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Bosch-Marce, M.; Okuyama, H.; Krishnamachary, B.; Kimura, H.; Zhang, L.; Huso, D.L.; Semenza, G.L. Spontaneous transformation of cultured mouse bone marrow–derived stromal cells. Cancer Res. 2006, 66, 10849–10854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.; Geissler, E.; Schlitt, H.; Baan, C.C.; Dahlke, M.H.; Hoogduijn, M.J. Mesenchymal stem cells are short–lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Klinkhammer, B.M.; Kramann, R.; Mallau, M.; Makowska, A.; van Roeyen, C.R.; Rong, S.; Buecher, E.B.; Boor, P.; Kovacova, K.; et al. Mesenchymal stem cells from rats with chronic kidney disease exhibit premature senescence and loss of regenerative potential. PLoS ONE 2014, 9, e92115. [Google Scholar]
- Mias, C.; Trouche, E.; Seguelas, M.-H.; Calcagno, F.; Dignat-George, F.; Sabatier, F.; Piercecchi-Marti, M.-D.; Daniel, L.; Bianchi, P.; Calise, D.; et al. Ex Vivo pretreatment with melatonin improves survival, proangiogenic/mitogenic activity, and efficiency of mesenchymal stem cells injected into ischemic kidney. Stem Cells 2008, 26, 1749–1757. [Google Scholar] [CrossRef]
- Masoud, M.S.; Anwar, S.S.; Afzal, M.Z.; Mehmood, A.; Khan, S.N.; Riazuddin, S. Pre-conditioned mesenchymal stem cells ameliorate renal ischemic injury in rats by augmented survival and engraftment. J. Transl. Med. 2012, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Jung, S.K.; Noh, H.; Lee, S.H. Melatonin protects chronic kidney disease mesenchymal stem cells against senescence via PrPC–dependent enhancement of the mitochondrial function. J. Pineal Res. 2019, 66, e12535. [Google Scholar] [CrossRef] [Green Version]
- Haider, H.K.; Jiang, S.; Idris, N.M.; Ashraf, M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1α/CXCR4 signaling to promote myocardial repair. Circ. Res. 2008, 103, 1300–1308. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhang, Z.; Guo, J.; Ni, A.; Deb, A.; Zhang, L.; Mirotsou, M.; Pratt, R.E.; Dzau, V.J. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res. 2010, 106, 1753–1762. [Google Scholar] [CrossRef]
- Hu, X.; Yu, S.P.; Fraser, J.L.; Lu, Z.; Ogle, M.E.; Wang, J.-A.; Wei, L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 2008, 135, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Gnecchi, M.; He, H.; Liang, O.D.; Melo, L.G.; Morello, F.; Mu, H.; Noiseux, N.; Zhang, L.; Pratt, R.E.; Ingwall, J.S.; et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 2005, 11, 367–368. [Google Scholar] [CrossRef]
- Gao, F.; He, T.; Wang, H.; Yu, S.; Yi, D.; Liu, W.; Cai, Z. A promising strategy for the treatment of ischemic heart disease: Mesenchymal stem cell-mediated vascular endothelial growth factor gene transfer in rats. Can. J. Cardiol. 2007, 23, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tan, J.; Wang, Y.; Meldrum, K.K.; Dinarello, C.A.; Meldrum, D.R. IL–18 binding protein–expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction. Proc. Natl. Acad. Sci. USA 2009, 106, 17499–17504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Ma, N.; Ong, L.-L.; Nesselmann, C.; Klopsch, C.; Ladilov, Y.; Furlani, D.; Piechacek, C.; Moebius, J.M.; Lützow, K.; et al. Bcl–2 engineered MSCs inhibited apoptosis and improved heart function. Stem Cells 2007, 25, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.-H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal stem cells deliver exogenous microRNA–let7c via exosomes to attenuate renal fibrosis. Mol. Therap. 2016, 24, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Yu, X.; Zhang, B.; Zhang, H.; Fang, Y.; Liu, S.; Liu, T.; Ding, X. Atorvastatin improves survival of implanted stem cells in a rat model of renal ischemia-reperfusion injury. Am. J. Nephrol. 2014, 39, 466–475. [Google Scholar] [CrossRef]
- Altun, B.; Yilmaz, R.; Aki, T.; Akoglu, H.; Zeybek, D.; Piskinpasa, S.; Uckan, D.; Purali, N.; Korkusuz, P.; Turgan, C. Use of mesenchymal stem cells and darbepoetin improve ischemia–induced acute kidney injury outcomes. Am. J. Nephrol. 2012, 35, 531–539. [Google Scholar] [CrossRef]
- Bathgate, R.A.D.; Hsueh, A.J.; Sherwood, O.D. Physiology and Molecular Biology of the Relaxin Peptide Family; Elsevier: Amsterdam, The Netherlands, 2006; pp. 679–768. [Google Scholar]
- Samuel, C.S.; Hewitson, T.D. Relaxin in cardiovascular and renal disease. Kidney Int. 2006, 69, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Conrad, K.P.; von Versen–Höynck, F.; Baker, V.L. Potential role of the corpus luteum in maternal cardiovascular adaptation to pregnancy and preeclampsia risk. Am. J. Obstet. Gynecol. 2021, 226, 683–699. [Google Scholar] [CrossRef]
- Samuel, C.S.; Hewitson, T.D. Relaxin and the progression of kidney disease. Curr. Opin. Nephrol. Hypertens. 2009, 18, 9–14. [Google Scholar] [CrossRef]
- Bathgate, R.A.; Ivell, R.; Sanborn, B.M.; Sherwood, O.D.; Summers, R.J. International Union of Pharmacology LVII: Recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol. Rev. 2006, 58, 7–31. [Google Scholar] [CrossRef]
- Samuel, C.S.; Tian, H.; Zhao, L.; Amento, E.P. Relaxin is a key mediator of prostate growth and male reproductive tract development. Lab. Investig. 2003, 83, 1055–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood, O.D. Relaxin’s physiological roles and other diverse actions. Endocr. Rev. 2004, 25, 205–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, C.S.; Zhao, C.; Bathgate, R.A.D.; Du, X.J.; Summers, R.J.; Amento, E.P.; Walker, L.L.; McBurnie, M.; Zhao, L.; Treegar, G.W. The relaxin gene-knockout mouse: A model of progressive fibrosis. Ann. N. Y. Acad. Sci. 2005, 1041, 73–81. [Google Scholar] [CrossRef]
- Samuel, C.S.; Zhao, C.; Bond, C.P.; Hewitson, T.D.; Amento, E.P.; Summers, R.J. Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 2004, 65, 2054–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitson, T.D.; Zhao, C.; Wigg, B.; Lee, S.W.; Simpson, E.R.; Boon, W.C.; Samuel, C.S. Relaxin and castration in male mice protect from, but testosterone exacerbates, age-related cardiac and renal fibrosis, whereas estrogens are an independent determinant of organ size. Endocrinology 2012, 153, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stults, J.T.; Bourell, J.H.; Canova–Davis, E.; Ling, V.T.; Laramee, G.R.; Winslow, J.W.; Griffin, P.R.; Rinderknecht, E.; Vandlen, R.L. Structural characterization by mass spectrometry of native and recombinant human relaxin. Biomed. Environ. Mass Spectrom. 1990, 19, 655–664. [Google Scholar] [CrossRef]
- Garber, S.L.; Mirochnik, Y.; Brecklin, C.; Slobodskoy, L.; Arruda, J.A.; Dunea, G. Effect of relaxin in two models of renal mass reduction. Am. J. Nephrol. 2003, 23, 8–12. [Google Scholar] [CrossRef]
- Danielson, L.A.; Welford, A.; Harris, A. Relaxin improves renal function and histology in aging munich wistar rats. J. Am. Soc. Nephrol. 2006, 17, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Sasser, J.M.; Cunningham, M.W., Jr.; Baylis, C. Serelaxin reduces oxidative stress and asymmetric dimethylarginine in angiotensin II–induced hypertension. Am. J. Physiol. Renal Physiol. 2014, 307, F1355–F1362. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.; Royce, S.; Hewitson, T.; Denton, K.; Cooney, T.; Bennett, R. Anti-fibrotic actions of relaxin. Br. J. Pharmacol. 2017, 174, 962–976. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Luo, Y.; Myakala, K.; Orlicky, D.J.; Dobrinskikh, E.; Wang, X.; Levi, M. Serelaxin improves cardiac and renal function in DOCA–salt hypertensive rats. Sci. Rep. 2017, 7, 9793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, S.L.; Mirochnik, Y.; Brecklin, C.S.; Unemori, E.N.; Singh, A.K.; Slobodskoy, L.; Grove, B.H.; Arruda, J.A.; Dunea, G. Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int. 2001, 59, 876–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
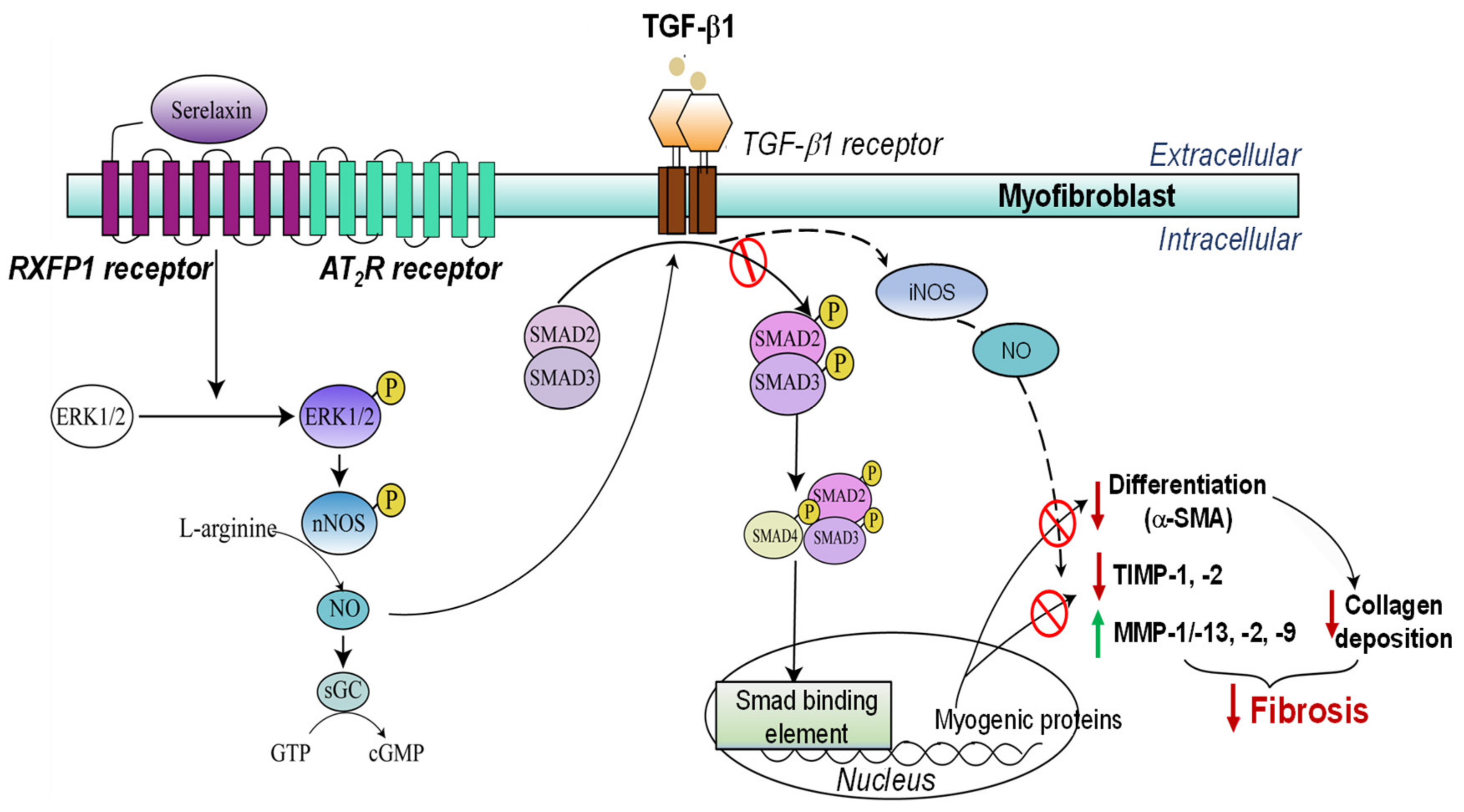
- Chow, B.S.M.; Kocan, M.; Shen, M.; Wang, Y.; Han, L.; Chew, J.Y.; Wang, C.; Bosnyak, S.; Mirabito-Colafella, K.M.; Barsha, G.; et al. AT1R-AT2R-RXFP1 functional crosstalk in myofibroblasts: Impact on the therapeutic targeting of renal and cardiac fibrosis. J. Am. Soc. Nephrol. 2019, 30, 2191–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Pinar, A.A.; Widdop, R.E.; Hossain, M.A.; Bathgate, R.A.; Denton, K.M.; Kemp-Harper, B.K.; Samuel, C.S. The anti-fibrotic actions of relaxin are mediated through AT2R-associated protein phosphatases via RXFP1-AT2R functional crosstalk in human cardiac myofibroblasts. FASEB J. 2020, 34, 8217–8233. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, I.; Hewitson, T.D.; Halls, M.L.; Summers, R.J.; Mathai, M.L.; Bathgate, R.A.D.; Tregear, G.W.; Samuel, C.S. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009, 23, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeg, M.H.J.; Koziolek, M.J.; Vasko, R.; Schaefer, L.; Sharma, K.; Müller, G.A.; Strutz, F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005, 68, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, M.; Samuel, C.S.; Bathgate, R.A.D.; Stewart, D.R.; Summers, R.J. Serelaxin-mediated signal transduction in human vascular cells: Bell-shaped concentration-response curves reflect differential coupling to G proteins. Br. J. Pharmacol. 2015, 172, 1005–1019. [Google Scholar] [CrossRef] [Green Version]
- Chow, B.S.M.; Chew, E.G.Y.; Zhao, C.; Bathgate, R.A.D.; Hewitson, T.D.; Samuel, C.S. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: The additional involvement of iNOS. PLoS ONE 2012, 7, e42714. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kemp–Harper, B.K.; Kocan, M.; Ang, S.Y.; Hewitson, T.D.; Samuel, C.S. The anti-fibrotic actions of relaxin are mediated through a NO-sGC-cGMP-dependent pathway in renal myofibroblasts in vitro and enhanced by the NO donor, diethylamine NONOate. Front. Pharmacol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Jeyabalan, A.; Conrad, K.P. Renal function during normal pregnancy and preeclampsia. Front. Biosci. 2007, 12, 2425–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathgate, R.A.D.; Halls, M.L.; van der Westhuizen, E.T.; Callander, G.E.; Kocan, M.; Summers, R.J. Relaxin family peptides and their receptors. Physiol. Rev. 2013, 93, 405–480. [Google Scholar] [CrossRef] [PubMed]
- Bani, D.; Masini, E.; Bello, M.G.; Bigazzi, M.; Sacchi, T.B. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am. J. Path. 1998, 152, 1367–1376. [Google Scholar] [PubMed]
- Masini, E.; Nistri, S.; Vannacci, A.; Sacchi, T.B.; Novelli, A.; Bani, D. Relaxin inhibits the activation of human neutrophils: Involvement of the nitric oxide pathway. Endocrinology 2004, 145, 1106–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-L.; Leu, J.-G.; Liu, W.-C.; Zheng, C.-M.; Lin, Y.-F.; Shyu, J.-F.; Wu, C.-C.; Lu, K.-C. Endothelial progenitor cells predict long–term mortality in hemodialysis patients. Int. J. Med. Sci. 2016, 13, 240. [Google Scholar] [CrossRef] [Green Version]
- Badawi, A.; Jefferson, O.C.; Huuskes, B.M.; Ricardo, S.D.; Kerr, P.G.; Samuel, C.S.; Murthi, P. A novel approach to enhance the regenerative potential of circulating endothelial progenitor cells in patients with end-stage kidney disease. Biomedicines 2022, 10, 883. [Google Scholar] [CrossRef]
- Tögel, F.; Hu, Z.; Weiss, K.; Isaac, J.; Lange, C.; Westenfelder, C. Administered mesenchymal stem cells protect 980 against ischemic acute renal failure through differentiation-independent mechanisms. Am. J. Physiol. Renal Physiol. 2005, 289, F31–F42. [Google Scholar] [CrossRef] [Green Version]
- Wise, A.F.; Williams, T.M.; Kiewiet, M.B.G.; Payne, N.L.; Siatskas, C.; Samuel, C.S.; Ricardo, S.D. Human mesenchymal stem cells alter macrophage phenotype and promote regeneration via homing to the kidney following ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1222–F1235. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Ricardo, S.D.; Samuel, C.S. Enhancing the Therapeutic Potential of Mesenchymal Stromal Cell-Based Therapies with an Anti-Fibrotic Agent for the Treatment of Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 6035. https://doi.org/10.3390/ijms23116035
Li Y, Ricardo SD, Samuel CS. Enhancing the Therapeutic Potential of Mesenchymal Stromal Cell-Based Therapies with an Anti-Fibrotic Agent for the Treatment of Chronic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(11):6035. https://doi.org/10.3390/ijms23116035
Chicago/Turabian StyleLi, Yifang, Sharon D. Ricardo, and Chrishan S. Samuel. 2022. "Enhancing the Therapeutic Potential of Mesenchymal Stromal Cell-Based Therapies with an Anti-Fibrotic Agent for the Treatment of Chronic Kidney Disease" International Journal of Molecular Sciences 23, no. 11: 6035. https://doi.org/10.3390/ijms23116035
APA StyleLi, Y., Ricardo, S. D., & Samuel, C. S. (2022). Enhancing the Therapeutic Potential of Mesenchymal Stromal Cell-Based Therapies with an Anti-Fibrotic Agent for the Treatment of Chronic Kidney Disease. International Journal of Molecular Sciences, 23(11), 6035. https://doi.org/10.3390/ijms23116035