
Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
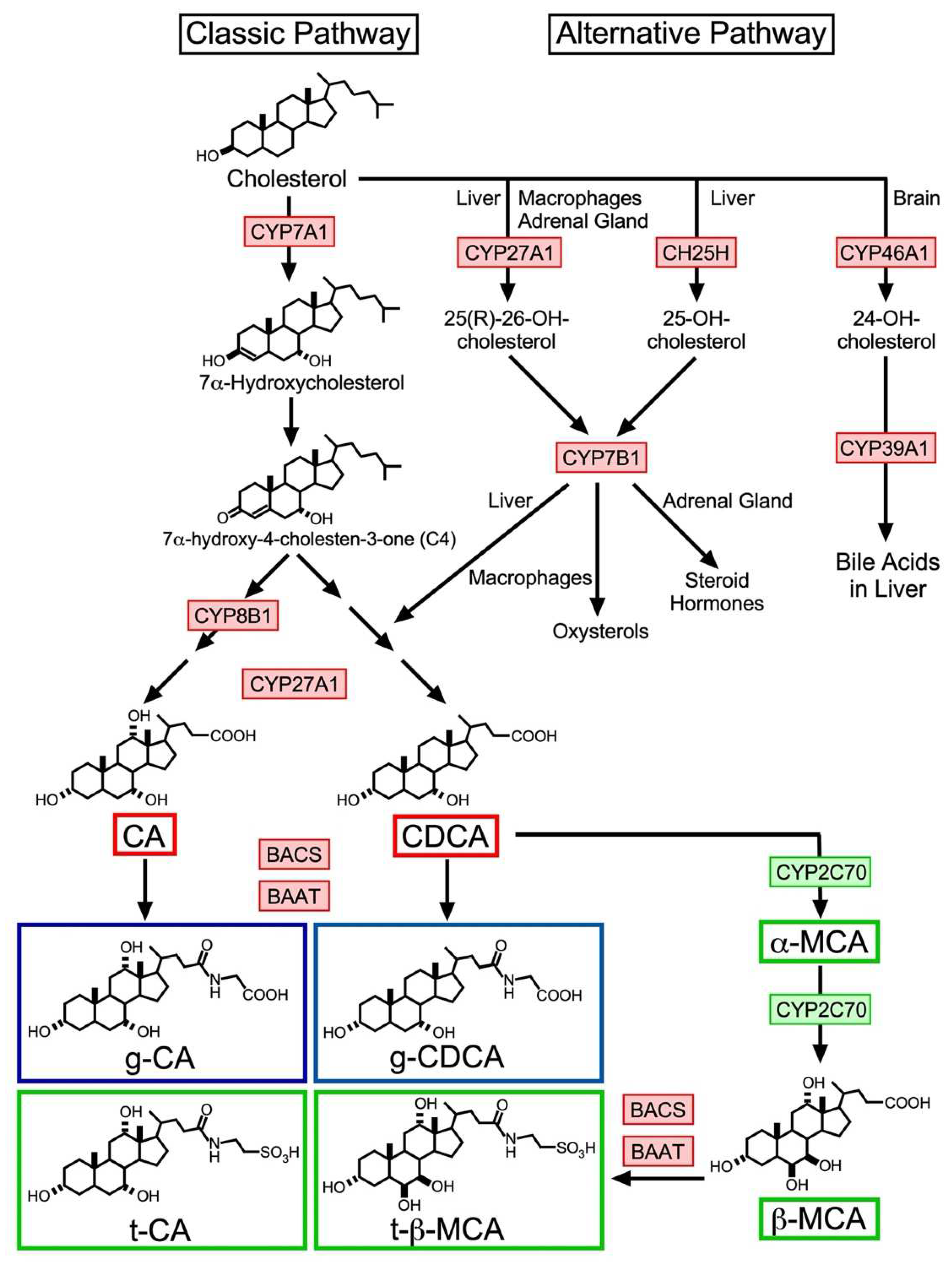
2. BA Biosynthesis
2.1. Classic Pathway
2.2. Alternative Pathway
2.3. Metabolism of BA by Gut Microbiota
2.4. Recycling of BA
3. FXR
3.1. FXR Structure
3.2. Liver- and Ileum-Specific Function of FXR
4. Regulation, Production and Biological Function of FGF15/19
4.1. Discovery of FGF15/19
4.2. Molecular Biological Basis for FXR-Dependent Regulation of FGF15/19
4.3. Interaction of FGF15/19 with FGFR4-KLβ Heterodimer
4.4. Regulation of CYP7A1 Expression by FGF15/19
4.5. Regulation of Other Biological Events by FGF15/19
4.6. Non-Hepatic Targets of FGF15/19
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Sheikh Abdul Kadir, S.H.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov, A.V.; Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef]
- Xie, M.H.; Holcomb, I.; Deuel, B.; Dowd, P.; Huang, A.; Vagts, A.; Foster, J.; Liang, J.; Brush, J.; Gu, Q.; et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 1999, 11, 729–735. [Google Scholar] [CrossRef]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro, O.M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ge, H.; Gupte, J.; Weiszmann, J.; Shimamoto, G.; Stevens, J.; Hawkins, N.; Lemon, B.; Shen, W.; Xu, J.; et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J. Biol. Chem. 2007, 282, 29069–29072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Li, G.; Thomas, A.M.; Hart, S.N.; Zhong, X.; Wu, D.; Guo, G.L. Farnesoid X receptor activation mediates head-to-tail chromatin looping in the Nr0b2 gene encoding small heterodimer partner. Mol. Endocrinol. 2010, 24, 1404–1412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Feng, S.; Li, J.; Wu, Z.; Deng, Q.; Yang, W.; Li, J.; Pan, G. Farnesoid X receptor (FXR) agonists induce hepatocellular apoptosis and impair hepatic functions via FXR/SHP pathway. Arch. Toxicol. 2022, 96, 1829–1843. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Baghdasaryan, A.; Claudel, T.; Gumhold, J.; Silbert, D.; Adorini, L.; Roda, A.; Vecchiotti, S.; Gonzalez, F.J.; Schoonjans, K.; Strazzabosco, M.; et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/-(Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO−₃ output. Hepatology 2011, 54, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Seok, S.; Zhang, Y.; Ma, J.; Kong, B.; Guo, G.; Kemper, B.; Kemper, J.K. Intestinal FGF15/19 physiologically repress hepatic lipogenesis in the late fed-state by activating SHP and DNMT3A. Nat. Commun. 2020, 11, 5969. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Damron, H.A.; Hillgartner, F.B. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 2009, 284, 10023–10033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Kim, Y.C.; Zhang, Y.; Kong, B.; Guo, G.; Sadoshima, J.; Ma, J.; Kemper, B.; Kemper, J.K. A postprandial FGF19-SHP-LSD1 regulatory axis mediates epigenetic repression of hepatic autophagy. EMBO J. 2017, 36, 1755–1769. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F. The function of bile salts in fat absorption. The solvent properties of dilute micellar solutions of conjugated bile salts. Biochem. J. 1963, 89, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Duane, W.C.; Javitt, N.B. 27-hydroxycholesterol: Production rates in normal human subjects. J. Lipid Res. 1999, 40, 1194–1199. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Investig. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Erickson, S.K.; Lear, S.R.; Deane, S.; Dubrac, S.; Huling, S.L.; Nguyen, L.; Bollineni, J.S.; Shefer, S.; Hyogo, H.; Cohen, D.E.; et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7A1-deficient mice. J. Lipid Res. 2003, 44, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Gälman, C.; Arvidsson, I.; Angelin, B.; Rudling, M. Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J. Lipid Res. 2003, 44, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, A.; Yoshida, T.; Xu, G.; Matsuzaki, Y.; Fukushima, S.; Tanaka, N.; Doy, M.; Shefer, S.; Salen, G. Significance of plasma 7alpha-hydroxy-4-cholesten-3-one and 27-hydroxycholesterol concentrations as markers for hepatic bile acid synthesis in cholesterol-fed rabbits. Metabolism 2004, 53, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y.L.; Ferrell, J.M. Targeting the gut microbiota for treating colitis: Is FGF19 a magic bullet? EBioMedicine 2020, 55, 102754. [Google Scholar] [CrossRef]
- Ali, Z.; Heverin, M.; Olin, M.; Acimovic, J.; Lövgren-Sandblom, A.; Shafaati, M.; Båvner, A.; Meiner, V.; Leitersdorf, E.; Björkhem, I. On the regulatory role of side-chain hydroxylated oxysterols in the brain. Lessons from CYP27A1 transgenic and Cyp27a1(-/-) mice. J. Lipid Res. 2013, 54, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Kakiyama, G.; Marques, D.; Takei, H.; Nittono, H.; Erickson, S.; Fuchs, M.; Rodriguez-Agudo, D.; Gil, G.; Hylemon, P.B.; Zhou, H.; et al. Mitochondrial oxysterol biosynthetic pathway gives evidence for CYP7B1 as controller of regulatory oxysterols. J. Steroid Biochem. Mol. Biol. 2019, 189, 36–47. [Google Scholar] [CrossRef]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef] [Green Version]
- Meaney, S.; Bodin, K.; Diczfalusy, U.; Bjorkhem, I. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: Critical importance of the position of the oxygen function. J. Lipid Res. 2002, 43, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- Babiker, A.; Diczfalusy, U. Transport of side-chain oxidized oxysterols in the human circulation. Biochim. Biophys. Acta 1998, 1392, 333–339. [Google Scholar] [CrossRef]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.; Shah, R.; Kotti, T. Cholesterol 24-hydroxylase: An enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkhem, I.; Lütjohann, D.; Diczfalusy, U.; Ståhle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar] [CrossRef]
- Rizzolo, D.; Buckley, K.; Kong, B.; Zhan, L.; Shen, J.; Stofan, M.; Brinker, A.; Goedken, M.; Buckley, B.; Guo, G.L. Bile acid homeostasis in a cholesterol 7α-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology 2019, 70, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, D.; Kong, B.; Taylor, R.E.; Brinker, A.; Goedken, M.; Buckley, B.; Guo, G.L. Bile acid homeostasis in female mice deficient in Cyp7a1 and Cyp27a1. Acta Pharm. Sin. B 2021, 11, 3847–3856. [Google Scholar] [CrossRef]
- Berr, F.; Kullak-Ublick, G.A.; Paumgartner, G.; Münzing, W.; Hylemon, P.B. 7 alpha-dehydroxylating bacteria enhance deoxycholic acid input and cholesterol saturation of bile in patients with gallstones. Gastroenterology 1996, 111, 1611–1620. [Google Scholar] [CrossRef]
- Hu, X.; Bonde, Y.; Eggertsen, G.; Rudling, M. Muricholic bile acids are potent regulators of bile acid synthesis via a positive feedback mechanism. J. Intern. Med. 2014, 275, 27–38. [Google Scholar] [CrossRef]
- Setchell, K.D.; Lawson, A.M.; Tanida, N.; Sjövall, J. General methods for the analysis of metabolic profiles of bile acids and related compounds in feces. J. Lipid Res. 1983, 24, 1085–1100. [Google Scholar] [CrossRef]
- Marion, S.; Desharnais, L.; Studer, N.; Dong, Y.; Notter, M.D.; Poudel, S.; Menin, L.; Janowczyk, A.; Hettich, R.L.; Hapfelmeier, S.; et al. Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res. 2020, 61, 1450–1463. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Physiological role of bile acids modified by the gut microbiome. Microorganisms 2021, 10, 68. [Google Scholar] [CrossRef]
- Guzior, D.V.; Quinn, R.A. Review: Microbial transformations of human bile acids. Microbiome 2021, 9, 140. [Google Scholar] [CrossRef]
- Daly, J.W.; Keely, S.J.; Gahan, C.G.M. Functional and phylogenetic diversity of BSH and PVA enzymes. Microorganisms 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, N.; Desharnais, L.; Beutler, M.; Brugiroux, S.; Terrazos, M.A.; Menin, L.; Schürch, C.M.; McCoy, K.D.; Kuehne, S.A.; Minton, N.P.; et al. Functional intestinal bile acid 7α-dehydroxylation by clostridium scindens associated with protection from clostridium difficile infection in a gnotobiotic mouse model. Front. Cell. Infect. Microbiol. 2016, 6, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepercq, P.; Gérard, P.; Béguet, F.; Raibaud, P.; Grill, J.P.; Relano, P.; Cayuela, C.; Juste, C. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol. Lett. 2004, 235, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce Τ(H)17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Alexander, M.; Ang, Q.Y.; Nayak, R.R.; Bustion, A.E.; Sandy, M.; Zhang, B.; Upadhyay, V.; Pollard, K.S.; Lynch, S.V.; Turnbaugh, P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022, 30, 17–30.e9. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Li, Y.; Cao, S.; Zhang, Y.; Wang, Z.; Liu, G.; Li, J.; Gu, B. Apical sodium-dependent bile acid transporter, drug target for bile acid related diseases and delivery target for prodrugs: Current and future challenges. Pharmacol. Ther. 2020, 212, 107539. [Google Scholar] [CrossRef]
- Oelkers, P.; Kirby, L.C.; Heubi, J.E.; Dawson, P.A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Investig. 1997, 99, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Shneider, B.L.; Dawson, P.A.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Investig. 1995, 95, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrefai, W.A.; Gill, R.K. Bile acid transporters: Structure, function, regulation and pathophysiological implications. Pharm. Res. 2007, 24, 1803–1823. [Google Scholar] [CrossRef]
- Praslickova, D.; Torchia, E.C.; Sugiyama, M.G.; Magrane, E.J.; Zwicker, B.L.; Kolodzieyski, L.; Agellon, L.B. The ileal lipid binding protein is required for efficient absorption and transport of bile acids in the distal portion of the murine small intestine. PLoS ONE 2012, 7, e50810. [Google Scholar] [CrossRef] [Green Version]
- Ballatori, N.; Fang, F.; Christian, W.V.; Li, N.; Hammond, C.L. Ostalpha-Ostbeta is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 295, G179–G186. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Hubbert, M.L.; Rao, A. Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim. Biophys. Acta 2010, 1801, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Kullak-Ublick, G.A.; Stieger, B.; Hagenbuch, B.; Meier, P.J. Hepatic transport of bile salts. Semin. Liver Dis. 2000, 20, 273–292. [Google Scholar] [CrossRef] [Green Version]
- Slijepcevic, D.; Roscam Abbing, R.L.P.; Katafuchi, T.; Blank, A.; Donkers, J.M.; van Hoppe, S.; de Waart, D.R.; Tolenaars, D.; van der Meer, J.H.M.; Wildenberg, M.; et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 2017, 66, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.L.; Köhler, H.; Röthlisberger, B.; Grobholz, R.; McLin, V.A. Sodium taurocholate co-transporting polypeptide deficiency. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101824. [Google Scholar] [CrossRef]
- Suga, T.; Yamaguchi, H.; Sato, T.; Maekawa, M.; Goto, J.; Mano, N. Preference of conjugated bile acids over unconjugated bile acids as substrates for OATP1B1 and OATP1B3. PLoS ONE 2017, 12, e0169719. [Google Scholar] [CrossRef]
- Ben Saad, A.; Bruneau, A.; Mareux, E.; Lapalus, M.; Delaunay, J.L.; Gonzales, E.; Jacquemin, E.; Aït-Slimane, T.; Falguières, T. Molecular regulation of canalicular ABC transporters. Int. J. Mol. Sci. 2021, 22, 2113. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.H.; Portincasa, P. Bile acid physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid X receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Carotti, A.; Marinozzi, M.; Custodi, C.; Cerra, B.; Pellicciari, R.; Gioiello, A.; Macchiarulo, A. Beyond bile acids: Targeting Farnesoid X Receptor (FXR) with natural and synthetic ligands. Curr. Top. Med. Chem. 2014, 14, 2129–2142. [Google Scholar] [CrossRef]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kast-Woelbern, H.R.; Edwards, P.A. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J. Biol. Chem. 2003, 278, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.M.; Murphy, K.; Miao, B.; Link, J.R.; Cunningham, M.R.; Rupar, M.J.; Gunyuzlu, P.L.; Haws, T.F.; Kassam, A.; Powell, F.; et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 2002, 290, 35–43. [Google Scholar] [CrossRef]
- Vaquero, J.; Monte, M.J.; Dominguez, M.; Muntané, J.; Marin, J.J. Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem. Pharmacol. 2013, 86, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Boesjes, M.; Bloks, V.W.; Hageman, J.; Bos, T.; van Dijk, T.H.; Havinga, R.; Wolters, H.; Jonker, J.W.; Kuipers, F.; Groen, A.K. Hepatic farnesoid X-receptor isoforms α2 and α4 differentially modulate bile salt and lipoprotein metabolism in mice. PLoS ONE 2014, 9, e115028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, J.C.; Massart, J.; de Boer, J.F.; Porsmyr-Palmertz, M.; Martínez-Redondo, V.; Agudelo, L.Z.; Sinha, I.; Meierhofer, D.; Ribeiro, V.; Björnholm, M.; et al. Bioenergetic cues shift FXR splicing towards FXRα2 to modulate hepatic lipolysis and fatty acid metabolism. Mol. Metab. 2015, 4, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Wärnmark, A.; Treuter, E.; Wright, A.P.; Gustafsson, J.A. Activation functions 1 and 2 of nuclear receptors: Molecular strategies for transcriptional activation. Mol. Endocrinol. 2003, 17, 1901–1909. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Ji, L.; Gu, J.; Zhou, M.; Chen, S. Farnesoid X receptor associates with β-catenin and inhibits its activity in hepatocellular carcinoma. Oncotarget 2015, 6, 4226–4238. [Google Scholar] [CrossRef] [Green Version]
- Zavacki, A.M.; Lehmann, J.M.; Seol, W.; Willson, T.M.; Kliewer, S.A.; Moore, D.D. Activation of the orphan receptor RIP14 by retinoids. Proc. Natl. Acad. Sci. USA 1997, 94, 7909–7914. [Google Scholar] [CrossRef] [Green Version]
- Laffitte, B.A.; Kast, H.R.; Nguyen, C.M.; Zavacki, A.M.; Moore, D.D.; Edwards, P.A. Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. J. Biol. Chem. 2000, 275, 10638–10647. [Google Scholar] [CrossRef] [Green Version]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [Green Version]
- Downes, M.; Verdecia, M.A.; Roecker, A.J.; Hughes, R.; Hogenesch, J.B.; Kast-Woelbern, H.R.; Bowman, M.E.; Ferrer, J.L.; Anisfeld, A.M.; Edwards, P.A.; et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol. Cell 2003, 11, 1079–1092. [Google Scholar] [CrossRef]
- Thomas, A.M.; Hart, S.N.; Kong, B.; Fang, J.; Zhong, X.B.; Guo, G.L. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2010, 51, 1410–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Liu, H.X.; Fang, Y.; Kong, B.; He, Y.; Zhong, X.B.; Fang, J.; Wan, Y.J.; Guo, G.L. Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS ONE 2014, 9, e105930. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Feng, X.; Rong, H.; Pan, Z.; Inaba, Y.; Qiu, L.; Zheng, W.; Lin, S.; Wang, R.; Wang, Z.; et al. The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat. Commun. 2013, 4, 1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, Y.; Jiang, J.; Zhang, S.; Li, S.; Shan, L.; Huang, J.; Zhang, W.; Li, H. Discovery of natural products as novel and potent FXR antagonists by virtual screening. Front. Chem. 2018, 6, 140. [Google Scholar] [CrossRef] [Green Version]
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef]
- Williams, S.; Bledsoe, R.K.; Collins, J.L.; Boggs, S.; Lambert, M.H.; Miller, A.B.; Moore, J.; McKee, D.D.; Moore, L.; Nichols, J.; et al. X-ray crystal structure of the liver X receptor beta ligand binding domain: Regulation by a histidine-tryptophan switch. J. Biol. Chem. 2003, 278, 27138–27143. [Google Scholar] [CrossRef] [Green Version]
- Mi, L.Z.; Devarakonda, S.; Harp, J.M.; Han, Q.; Pellicciari, R.; Willson, T.M.; Khorasanizadeh, S.; Rastinejad, F. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol. Cell 2003, 11, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Fujino, T.; Sato, Y.; Une, M.; Kanayasu-Toyoda, T.; Yamaguchi, T.; Shudo, K.; Inoue, K.; Nishimaki-Mogami, T. In vitro farnesoid X receptor ligand sensor assay using surface plasmon resonance and based on ligand-induced coactivator association. J. Steroid Biochem. Mol. Biol. 2003, 87, 247–252. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, S.; Gao, M.; Liu, J.; Jia, X.; Han, Q.; Zheng, S.; Miao, Y.; Li, S.; Weng, H.; et al. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 2277–2282. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Jia, P.; Fang, Y.; Jin, J.; Sun, Z.; Zhou, W.; Li, J.; Zhang, Y.; Wang, X.; Ren, T.; et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022, 101, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; van der Sluis, R.J.; Li, Z.; Oosterveer, M.H.; Groen, A.K.; Van Berkel, T.J. FXR agonist GW4064 increases plasma glucocorticoid levels in C57BL/6 mice. Mol. Cell. Endocrinol. 2012, 362, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Amar, M.J.; Guo, G.; Brewer, H.B., Jr.; Gonzalez, F.J.; Sinal, C.J. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 2003, 278, 2563–2570. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yin, L.; Anderson, J.; Ma, H.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J. Biol. Chem. 2010, 285, 3035–3043. [Google Scholar] [CrossRef] [Green Version]
- Welch, R.D.; Billon, C.; Losby, M.; Bedia-Diaz, G.; Fang, Y.; Avdagic, A.; Elgendy, B.; Burris, T.P.; Griffett, K. Emerging role of nuclear receptors for the treatment of NAFLD and NASH. Metabolites 2022, 12, 238. [Google Scholar] [CrossRef]
- Kremoser, C. FXR agonists for NASH: How are they different and what difference do they make? J. Hepatol. 2021, 75, 12–15. [Google Scholar] [CrossRef]
- Maliha, S.; Guo, G.L. Farnesoid X receptor and fibroblast growth factor 15/19 as pharmacological targets. Liver Res. 2021, 5, 142–150. [Google Scholar] [CrossRef]
- Hwang, S.T.; Urizar, N.L.; Moore, D.D.; Henning, S.J. Bile acids regulate the ontogenic expression of ileal bile acid binding protein in the rat via the farnesoid X receptor. Gastroenterology 2002, 122, 1483–1492. [Google Scholar] [CrossRef]
- Landrier, J.F.; Eloranta, J.J.; Vavricka, S.R.; Kullak-Ublick, G.A. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G476–G485. [Google Scholar] [CrossRef] [Green Version]
- Neimark, E.; Chen, F.; Li, X.; Shneider, B.L. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 2004, 40, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, G.; Shang, Q.; Pan, L.; Shefer, S.; Batta, A.K.; Bollineni, J.; Tint, G.S.; Keller, B.T.; Salen, G. Inhibition of ileal bile acid transport lowers plasma cholesterol levels by inactivating hepatic farnesoid X receptor and stimulating cholesterol 7 alpha-hydroxylase. Metabolism 2004, 53, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile acids as hormones: The FXR-FGF15/19 pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarrillo, C.M.; Keirns, B.H.; Koemel, N.A.; Anderson, K.L.; Emerson, S.R. Fibroblast growth factor 19: Potential modulation of hepatic metabolism for the treatment of non-alcoholic fatty liver disease. Liver Int. 2021, 41, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152. [Google Scholar] [CrossRef]
- Roberts, S.K.; Majeed, A. A short report on NGM282/aldafermin for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Ther. Targets 2021, 25, 889–895. [Google Scholar] [CrossRef]
- Lu, Q.; Wright, D.D.; Kamps, M.P. Fusion with E2A converts the Pbx1 homeodomain protein into a constitutive transcriptional activator in human leukemias carrying the t(1;19) translocation. Mol. Cell. Biol. 1994, 14, 3938–3948. [Google Scholar] [CrossRef]
- McWhirter, J.R.; Goulding, M.; Weiner, J.A.; Chun, J.; Murre, C. A novel fibroblast growth factor gene expressed in the developing nervous system is a downstream target of the chimeric homeodomain oncoprotein E2A-Pbx1. Development 1997, 124, 3221–3232. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 2008, 237, 18–27. [Google Scholar] [CrossRef]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Nishimura, T.; Utsunomiya, Y.; Hoshikawa, M.; Ohuchi, H.; Itoh, N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim. Biophys. Acta 1999, 1444, 148–151. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Evolutionary conservation of CCND1-ORAOV1-FGF19-FGF4 locus from zebrafish to human. Int. J. Mol. Med. 2003, 12, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; McMahon, A.P. A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development 2002, 129, 4807–4819. [Google Scholar] [CrossRef] [PubMed]
- Borello, U.; Cobos, I.; Long, J.E.; McWhirter, J.R.; Murre, C.; Rubenstein, J.L. FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev. 2008, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.J.; Ladher, R.; McWhirter, J.; Murre, C.; Schoenwolf, G.C.; Mansour, S.L. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev. Biol. 2004, 269, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Kurose, H.; Bito, T.; Adachi, T.; Shimizu, M.; Noji, S.; Ohuchi, H. Expression of Fibroblast growth factor 19 (Fgf19) during chicken embryogenesis and eye development, compared with Fgf15 expression in the mouse. Gene Expr. Patterns 2004, 4, 687–693. [Google Scholar] [CrossRef]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.P.; et al. A mouse model of hepatocellular carcinoma: Ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef]
- Maloney, P.R.; Parks, D.J.; Haffner, C.D.; Fivush, A.M.; Chandra, G.; Plunket, K.D.; Creech, K.L.; Moore, L.B.; Wilson, J.G.; Lewis, M.C.; et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 2000, 43, 2971–2974. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Holmstrom, S.R.; Fon Tacer, K.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J. Biol. Chem. 2010, 285, 14486–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukovac, S.; Los, E.L.; Stellaard, F.; Rings, E.H.; Verkade, H.J. Effects of essential fatty acid deficiency on enterohepatic circulation of bile salts in mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 297, G520–G531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Miyata, M.; Takamatsu, Y.; Kuribayashi, H.; Yamazoe, Y. Administration of ampicillin elevates hepatic primary bile acid synthesis through suppression of ileal fibroblast growth factor 15 expression. J. Pharmacol. Exp. Ther. 2009, 331, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Kuribayashi, H.; Miyata, M.; Yamakawa, H.; Yoshinari, K.; Yamazoe, Y. Enterobacteria-mediated deconjugation of taurocholic acid enhances ileal farnesoid X receptor signaling. Eur. J. Pharmacol. 2012, 697, 132–138. [Google Scholar] [CrossRef]
- Wahlström, A.; Kovatcheva-Datchary, P.; Ståhlman, M.; Khan, M.T.; Bäckhed, F.; Marschall, H.U. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 2017, 58, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Fujimori, T.; Furuya, A.; Satoh, J.; Nabeshima, Y.; Nabeshima, Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J. Clin. Investig. 2005, 115, 2202–2208. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, K.; Maeda, R.; Urakawa, I.; Yamazaki, Y.; Tanaka, T.; Ito, S.; Nabeshima, Y.; Tomita, T.; Odori, S.; Hosoda, K.; et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 1666–1671. [Google Scholar] [CrossRef] [Green Version]
- Katafuchi, T.; Esterházy, D.; Lemoff, A.; Ding, X.; Sondhi, V.; Kliewer, S.A.; Mirzaei, H.; Mangelsdorf, D.J. Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab. 2015, 21, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Wang, F.; Kan, M.; Jin, C.; Jones, R.B.; Weinstein, M.; Deng, C.X.; McKeehan, W.L. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol. Chem. 2000, 275, 15482–15489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.X.; Watts, L.M.; Manchem, V.P.; Chakravarty, K.; Monia, B.P.; McCaleb, M.L.; Bhanot, S. Peripheral reduction of FGFR4 with antisense oligonucleotides increases metabolic rate and lowers adiposity in diet-induced obese mice. PLoS ONE 2013, 8, e66923. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Zhao, J.; Wu, J.; Qiao, G.; Gu, J.; Zhou, C.; Li, Q.; Ying, L.; Wang, D.; Lin, H.; et al. Curtailing FGF19’s mitogenicity by suppressing its receptor dimerization ability. Proc. Natl. Acad. Sci. USA 2020, 117, 29025–29034. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The structural biology of the FGF19 subfamily. Adv. Exp. Med. Biol. 2012, 728, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.M.; Harper Calderon, J.; Hock, E.; Jimenez, Y.; Barringer, K.; Carbonaro, M.; Molina-Portela, M.D.P.; Thurston, G.; Li, Z.; Daly, C. Monomeric/dimeric forms of Fgf15/FGF19 show differential activity in hepatocyte proliferation and metabolic function. FASEB J. 2021, 35, e21286. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Lemon, B.; Vonderfecht, S.; Baribault, H.; Weiszmann, J.; Gupte, J.; Gardner, J.; Lindberg, R.; Wang, Z.; et al. Separating mitogenic and metabolic activities of fibroblast growth factor 19 (FGF19). Proc. Natl. Acad. Sci. USA 2010, 107, 14158–14163. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Kir, S.; Zhang, Y.; Gerard, R.D.; Kliewer, S.A.; Mangelsdorf, D.J. Nuclear receptors HNF4α and LRH-1 cooperate in regulating Cyp7a1 in vivo. J. Biol. Chem. 2012, 287, 41334–41341. [Google Scholar] [CrossRef] [Green Version]
- Ehrlund, A.; Treuter, E. Ligand-independent actions of the orphan receptors/corepressors DAX-1 and SHP in metabolism, reproduction and disease. J. Steroid Biochem. Mol. Biol. 2012, 130, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lee, Y.K.; Park, S.H.; Kim, Y.S.; Park, S.H.; Lee, J.W.; Kwon, H.B.; Soh, J.; Moore, D.D.; Choi, H.S. Structure and expression of the orphan nuclear receptor SHP gene. J. Biol. Chem. 1998, 273, 14398–14402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwinski, M.; Ni, H.M.; et al. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat. Commun. 2020, 11, 3612. [Google Scholar] [CrossRef]
- Qureshi, H.A.; Pearl, J.A.; Anderson, K.A.; Green, R.M. Fibroblast growth factor 19 activates the unfolded protein response and mitogen-activated protein kinase phosphorylation in H-69 cholangiocyte cells. J. Liver 2014, 3, 158. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; Choi, S.E.; Kim, D.H.; Seok, S.; Suino-Powell, K.M.; Xu, H.E.; Kemper, J.K. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor β-Klotho. Proc. Natl. Acad. Sci. USA 2012, 109, 16137–16142. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, H.; Chiba, T.; Ao, J.; Koroki, K.; Kanayama, K.; Maruta, S.; Maeda, T.; Kusakabe, Y.; Kobayashi, K.; Kanogawa, N.; et al. The impact of FGF19/FGFR4 signaling inhibition in antitumor activity of multi-kinase inhibitors in hepatocellular carcinoma. Sci. Rep. 2021, 11, 5303. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Lemon, B.; Vonderfecht, S.; Weiszmann, J.; Hecht, R.; Gupte, J.; Hager, T.; Wang, Z.; Lindberg, R.; et al. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J. Biol. Chem. 2010, 285, 5165–5170. [Google Scholar] [CrossRef] [Green Version]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 306, G893–G902. [Google Scholar] [CrossRef] [Green Version]
- Desnoyers, L.R.; Pai, R.; Ferrando, R.E.; Hötzel, K.; Le, T.; Ross, J.; Carano, R.; D’Souza, A.; Qing, J.; Mohtashemi, I.; et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008, 27, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Jiang, L.; Liang, L.; Koral, K.; Zhang, Q.; Zhao, L.; Lu, S.; Tao, J. The role of fibroblast growth factor 19 in hepatocellular carcinoma. Am. J. Pathol. 2021, 191, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lang, L.; Zhao, X.; Shay, C.; Shull, A.Y.; Teng, Y. FGF19 amplification reveals an oncogenic dependency upon autocrine FGF19/FGFR4 signaling in head and neck squamous cell carcinoma. Oncogene 2019, 38, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, H.; Liu, Z.; Li, K.; Qin, R.; Wang, Y.; Liu, J.; Li, Z.; Gao, Q.; Pan, C.; et al. FGF19 and FGFR4 promotes the progression of gallbladder carcinoma in an autocrine pathway dependent on GPBAR1-cAMP-EGR1 axis. Oncogene 2021, 40, 4941–4953. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Byun, S.; Zhang, Y.; Seok, S.; Kemper, B.; Ma, J.; Kemper, J.K. Liver ChIP-seq analysis in FGF19-treated mice reveals SHP as a global transcriptional partner of SREBP-2. Genome Biol. 2015, 16, 268. [Google Scholar] [CrossRef] [Green Version]
- Akalestou, E.; Miras, A.D.; Rutter, G.A.; le Roux, C.W. Mechanisms of weight loss after obesity surgery. Endocr. Rev. 2022, 43, 19–34. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.A.; Durieux, A.C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Marcelin, G.; Blouet, C.; Jeong, J.H.; Jo, Y.H.; Schwartz, G.J.; Chua, S., Jr. A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol. Metab. 2018, 8, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ursic-Bedoya, J.; Chavey, C.; Desandré, G.; Meunier, L.; Dupuy, A.M.; Gonzalez-Dopeso Reyes, I.; Tordjmann, T.; Assénat, E.; Hibner, U.; Gregoire, D. Fibroblast growth factor 19 stimulates water intake. Mol. Metab. 2022, 60, 101483. [Google Scholar] [CrossRef]
- Picard, A.; Metref, S.; Tarussio, D.; Dolci, W.; Berney, X.; Croizier, S.; Labouebe, G.; Thorens, B. Fgf15 neurons of the dorsomedial hypothalamus control glucagon secretion and hepatic gluconeogenesis. Diabetes 2021, 70, 1443–1457. [Google Scholar] [CrossRef]
- Picard, A.; Soyer, J.; Berney, X.; Tarussio, D.; Quenneville, S.; Jan, M.; Grouzmann, E.; Burdet, F.; Ibberson, M.; Thorens, B. A genetic screen identifies hypothalamic Fgf15 as a regulator of glucagon secretion. Cell Rep. 2016, 17, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Hultman, K.; Scarlett, J.M.; Baquero, A.F.; Cornea, A.; Zhang, Y.; Salinas, C.B.G.; Brown, J.; Morton, G.J.; Whalen, E.J.; Grove, K.L.; et al. The central fibroblast growth factor receptor/beta klotho system: Comprehensive mapping in Mus musculus and comparisons to nonhuman primate and human samples using an automated in situ hybridization platform. J. Comp. Neurol. 2019, 527, 2069–2085. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katafuchi, T.; Makishima, M. Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis. Int. J. Mol. Sci. 2022, 23, 6046. https://doi.org/10.3390/ijms23116046
Katafuchi T, Makishima M. Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis. International Journal of Molecular Sciences. 2022; 23(11):6046. https://doi.org/10.3390/ijms23116046
Chicago/Turabian StyleKatafuchi, Takeshi, and Makoto Makishima. 2022. "Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis" International Journal of Molecular Sciences 23, no. 11: 6046. https://doi.org/10.3390/ijms23116046
APA StyleKatafuchi, T., & Makishima, M. (2022). Molecular Basis of Bile Acid-FXR-FGF15/19 Signaling Axis. International Journal of Molecular Sciences, 23(11), 6046. https://doi.org/10.3390/ijms23116046