
Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis


Abstract
:1. Introduction
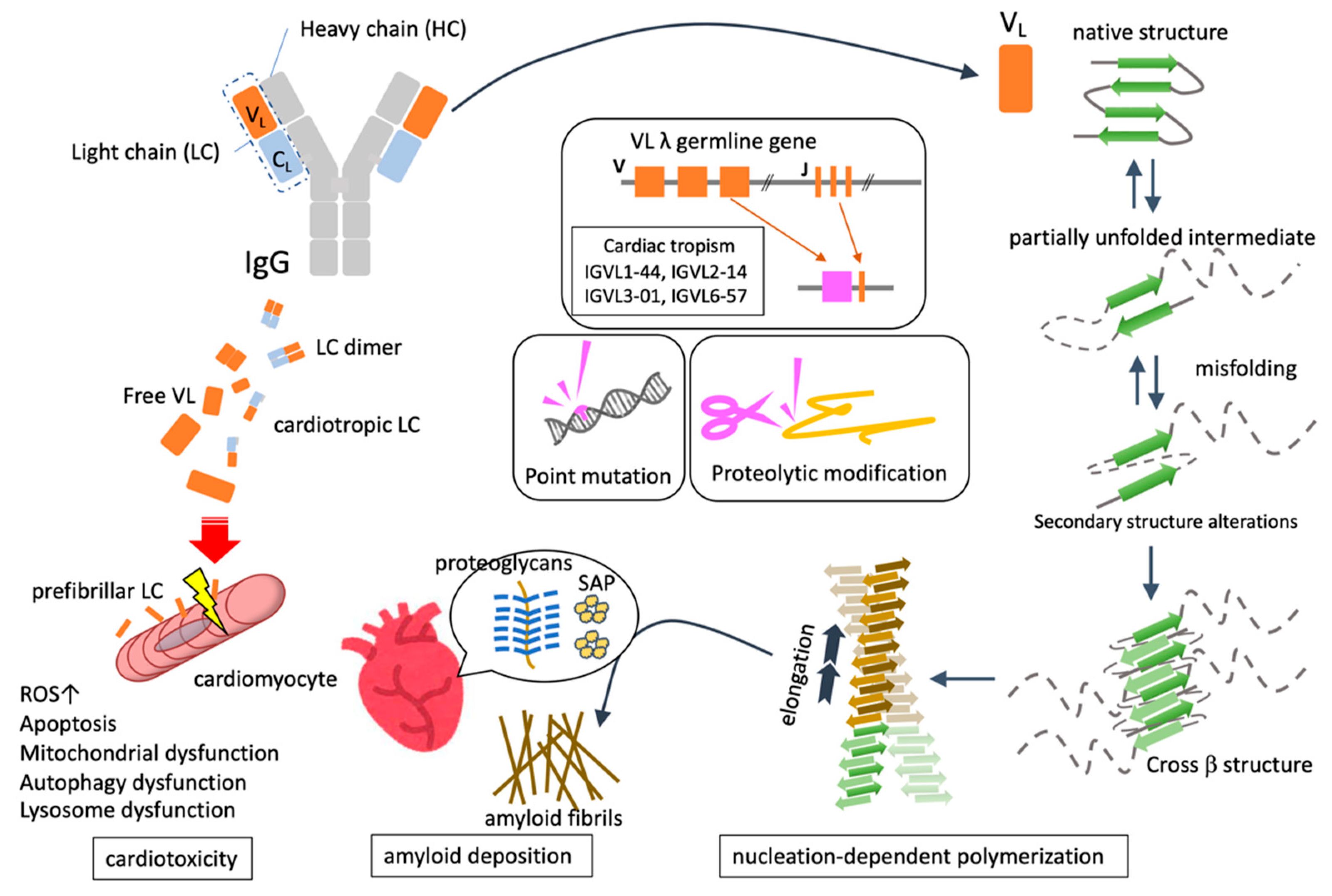
2. Pathogenesis of AL Amyloidosis
2.1. Common Mechanisms of Amyloid Fibril Formation
2.1.1. Protein Unfolding and Misfolding
2.1.2. Nucleation-Dependent Polymerization
2.1.3. Deposition of Amyloid Fibrils
3. Characteristics of AL Amyloidosis
3.1. FLC as a Precursor Protein of AL
3.2. Quantitative and Qualitative Abnormality of FLC
3.3. Genomic Mutation Associated with the Pathogenesis of AL Amyloidosis
3.4. Proteolytic Modification for Amyloid Fibril Formation
3.5. Structural Characteristics of AL Amyloid Fibril
3.6. Deposited Components Together with Amyloid Fibril
3.7. The Molecular Mechanism Underlying Cardiac Damage in AL Amyloidosis

{kind=link}
{kind=link}
| Author [Year] [Reference] | Species | Mechanisms of Myocardial Damage Directly Exerted by AL-LC |
|---|---|---|
| Diomede et al. [2014] [51] | C. elegans | Administration of LC extracted from patients with AL-CM induced a significant reduction in the lifespan and mitochondrial dysfunction |
| Diomede et al. [2017] [52] | C. elegans | LC purified from patients with severe cardiac involvement intrinsically generated high levels of ROS and when administered to C. elegans induced ROS production |
| Shi et al. [2010] [53] | Rat | Human AL-LC induced apoptosis in isolated adult rat cardiomyocytes by TAB-1-mediated autophosphorylation of p38-MAPK |
| Brenner et al. [2004] [55] | Rat | Human amyloid LC proteins alter cellular redox state in isolated cardiomyocytes, marked by an increase in intracellular reactive oxygen species and upregulation of the redox-sensitive protein, heme oxygenase-1 |
| Guan et al. [2014] [54] | Rat Zebrafish | Cardiac autophagy dysfunction triggered by lysosomal disorders based on decreased TFEB expression leads to cardiotoxicity of AL-LC |
| Mishra et al. [2019] [56] | Zebrafish | AL-LC-induced cardiac dysfunction, pericardial edema, and increased mortality improved with the administration of p38 inhibitors |
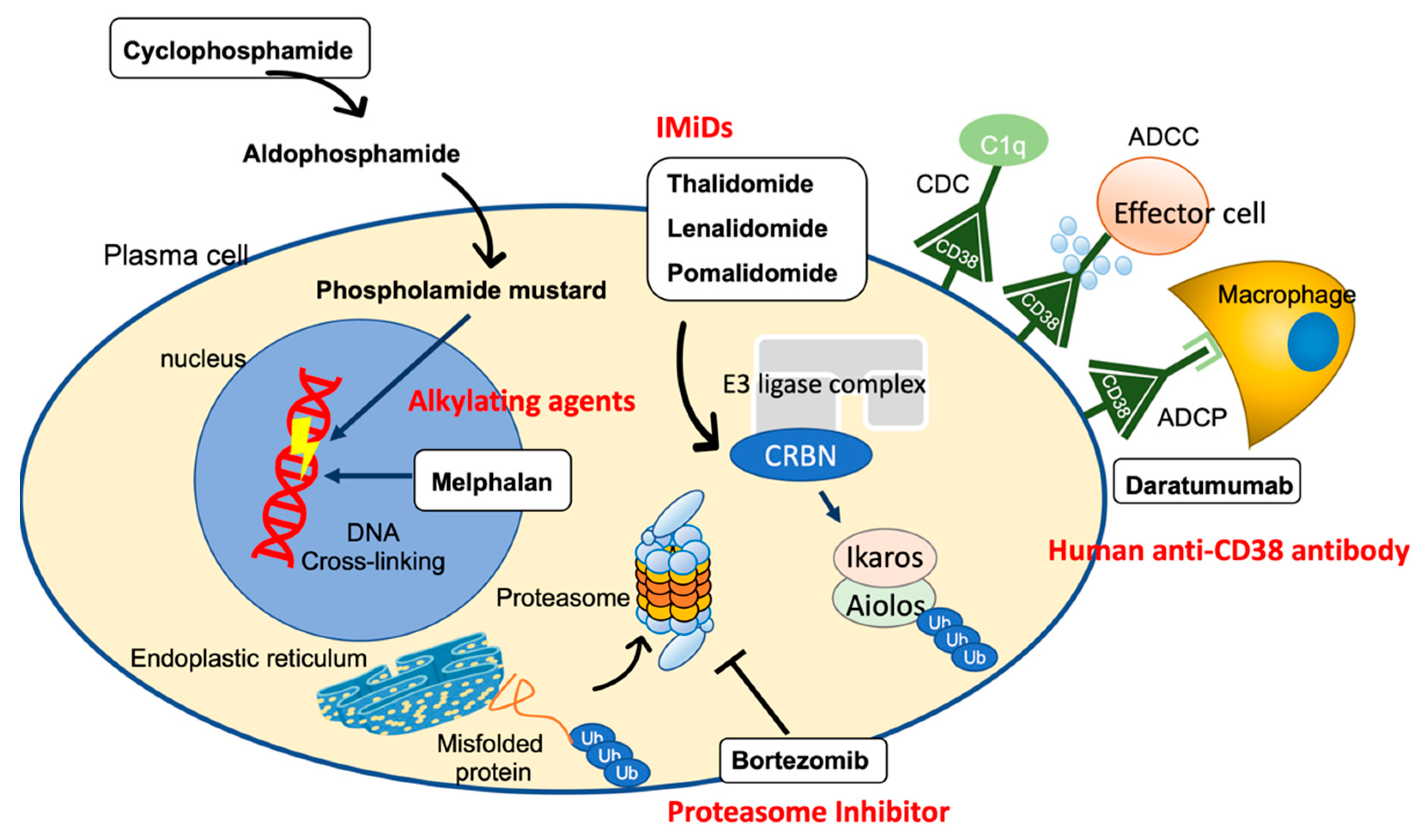
4. Treatment Strategies for AL Amyloidosis
4.1. Alkylating Agent
4.1.1. Melphalan
4.1.2. Cyclophosphamide
4.2. Proteasome Inhibitors (PIs)
4.3. Human Anti-CD38 Antibody (Daratumumab)
4.4. Immunomodulatory Drugs (IMiDs)
4.5. Drugs Targeting Amyloid Fibers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blake, C.C.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of Prealbumin: Secondary, Tertiary and Quaternary Interactions Determined by Fourier Refinement at 1.8 A. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a Complex of Two Plasma Proteins: Transthyretin and Retinol-Binding Protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Colon, W.; Kelly, J.W. Partial Denaturation of Transthyretin Is Sufficient for Amyloid Fibril Formation in Vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef]
- Brahmanandam, V.; McGraw, S.; Mirza, O.; Desai, A.A.; Farzaneh-Far, A. Regression of Cardiac Amyloidosis after Stem Cell Transplantation Assessed by Cardiovascular Magnetic Resonance Imaging. Circulation 2014, 129, 2326–2328. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.N. Serum Amyloid P Component Scintigraphy for Diagnosis and Monitoring Amyloidosis. Curr. Opin. Nephrol. Hypertens. 2002, 11, 649–655. [Google Scholar] [CrossRef]
- Falk, R.H.; Comenzo, R.L.; Skinner, M. The Systemic Amyloidoses. N. Engl. J. Med. 1997, 337, 898–909. [Google Scholar] [CrossRef]
- Merlini, G.; Stone, M.J. Dangerous Small B-Cell Clones. Blood J. Hematol. 2006, 108, 2520–2530. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Gertz, M.A. Primary Systemic Amyloidosis: Clinical and Laboratory Features in 474 Cases. Semin. Hematol. 1995, 32, 45–59. [Google Scholar]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The Tropism of Organ Involvement in Primary Systemic Amyloidosis: Contributions of Ig VL Germ Line Gene Use and Clonal Plasma Cell Burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Wechalekar, A.D.; Schonland, S.O.; Kastritis, E.; Gillmore, J.D.; Dimopoulos, M.A.; Lane, T.; Foli, A.; Foard, D.; Milani, P.; Rannigan, L.; et al. A European Collaborative Study of Treatment Outcomes in 346 Patients with Cardiac Stage III AL Amyloidosis. Blood J. Am. Soc. Hematol. 2013, 121, 3420–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaccard, A.; Moreau, P.; Leblond, V.; Leleu, X.; Benboubker, L.; Hermine, O.; Recher, C.; Asli, B.; Lioure, B.; Royer, B.; et al. High-Dose Melphalan versus Melphalan plus Dexamethasone for AL Amyloidosis. N. Engl. J. Med. 2007, 357, 1083–1093. [Google Scholar] [CrossRef]
- Comenzo, R.L.; Gertz, M.A. Autologous Stem Cell Transplantation for Primary Systemic Amyloidosis. Blood J. Hematol. 2002, 99, 4276–4282. [Google Scholar] [CrossRef]
- Skinner, M.; Sanchorawala, V.; Seldin, D.C.; Dember, L.M.; Falk, R.H.; Berk, J.L.; Anderson, J.J.; O’Hara, C.; Finn, K.T.; Libbey, C.A.; et al. High-Dose Melphalan and Autologous Stem-Cell Transplantation in Patients with AL Amyloidosis: An 8-Year Study. Ann. Intern. Med. 2004, 140, 85–93. [Google Scholar] [CrossRef]
- Kastritis, E.; Palladini, G.; Minnema, M.C.; Wechalekar, A.D.; Jaccard, A.; Lee, H.C.; Sanchorawala, V.; Gibbs, S.; Mollee, P.; Venner, C.P.; et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N. Engl. J. Med. 2021, 385, 46–58. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Khurana, R.; Gillespie, J.R.; Talapatra, A.; Minert, L.J.; Ionescu-Zanetti, C.; Millett, I.; Fink, A.L. Partially Folded Intermediates as Critical Precursors of Light Chain Amyloid Fibrils and Amorphous Aggregates. Biochemistry 2001, 40, 3525–3535. [Google Scholar] [CrossRef]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational Quality Control: Folding, Refolding, and Degrading Proteins. Science 1999, 286, 1888–1893. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Alvarado, M.; Kelly, J.W.; Dobson, C.M. Protein Misfolding Diseases: Current and Emerging Principles and Therapies; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Naiki, H.; Gejyo, F. Kinetic Analysis of Amyloid Fibril Formation. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1999; Volume 309, pp. 305–318. ISBN 0076-6879. [Google Scholar]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Bergström, J.; Gustavsson, A.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.-O.; Westermark, P. Amyloid Deposits in Transthyretin-Derived Amyloidosis: Cleaved Transthyretin Is Associated with Distinct Amyloid Morphology. J. Pathol. Bacteriol. 2005, 206, 224–232. [Google Scholar] [CrossRef]
- Stevens, F.J.; Kisilevsky, R. Immunoglobulin Light Chains, Glycosaminoglycans, and Amyloid. Cell. Mol. Life Sci. 2000, 57, 441–449. [Google Scholar] [CrossRef]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin Light Chain Amyloidosis—The Archetype of Structural and Pathogenic Variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying Immunoglobulin Gene Usage in Systemic and Localized Immunoglobulin Light-Chain Amyloidosis by Mass Spectrometry. Blood J. Am. Soc. Hematol. 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Perfetti, V.; Palladini, G.; Casarini, S.; Navazza, V.; Rognoni, P.; Obici, L.; Invernizzi, R.; Perlini, S.; Klersy, C.; Merlini, G. The Repertoire of λ Light Chains Causing Predominant Amyloid Heart Involvement and Identification of a Preferentially Involved Germline Gene, IGLV1-44. Blood J. Am. Soc. Hematol. 2012, 119, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Tischer, A.; Thompson, J.R.; Tai, J.; Wang, L.; Auton, M.; Ramirez-Alvarado, M. Kinetic Control in Protein Folding for Light Chain Amyloidosis and the Differential Effects of Somatic Mutations. J. Mol. Biol. 2014, 426, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazman, P.; Vielberg, M.-T.; Pulido Cendales, M.D.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal Amyloid Formation in a Patient’s Antibody Light Chain Is Caused by a Single Point Mutation. eLife 2020, 9, e52300. [Google Scholar] [CrossRef]
- Kim, Y.; Wall, J.S.; Meyer, J.; Murphy, C.; Randolph, T.W.; Manning, M.C.; Solomon, A.; Carpenter, J.F. Thermodynamic Modulation of Light Chain Amyloid Fibril Formation. J. Biol. Chem. 2000, 275, 1570–1574. [Google Scholar] [CrossRef] [Green Version]
- DiCostanzo, A.C.; Thompson, J.R.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M. Tyrosine Residues Mediate Fibril Formation in a Dynamic Light Chain Dimer Interface. J. Biol. Chem. 2012, 287, 27997–28006. [Google Scholar] [CrossRef] [Green Version]
- Misra, P.; Blancas-Mejia, L.M.; Ramirez-Alvarado, M. Mechanistic Insights into the Early Events in the Aggregation of Immunoglobulin Light Chains. Biochemistry 2019, 58, 3155–3168. [Google Scholar] [CrossRef]
- Röcken, C.; Stix, B.; Brömme, D.; Ansorge, S.; Roessner, A.; Bühling, F. A Putative Role for Cathepsin K in Degradation of AA and AL Amyloidosis. Am. J. Pathol. 2001, 158, 1029–1038. [Google Scholar] [CrossRef]
- Bohne, S.; Sletten, K.; Menard, R.; Bühling, F.; Vöckler, S.; Wrenger, E.; Roessner, A.; Röcken, C. Cleavage of AL Amyloid Proteins and AL Amyloid Deposits by Cathepsins B, K, and L. J. Pathol. Bacteriol. 2004, 203, 528–537. [Google Scholar] [CrossRef]
- Biolo, A.; Ramamurthy, S.; Connors, L.H.; O’Hara, C.J.; Meier-Ewert, H.K.; Soo Hoo, P.T.; Sawyer, D.B.; Seldin, D.C.; Seldin, D.S.; Sam, F. Matrix Metalloproteinases and Their Tissue Inhibitors in Cardiac Amyloidosis: Relationship to Structural, Functional Myocardial Changes and to Light Chain Amyloid Deposition. Circulation 2008, 1, 249–257, Correction in Circ. Heart Fail. 2009, 2, E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Essick, E.E.; Doros, G.; Tanriverdi, K.; Connors, L.H.; Seldin, D.C.; Sam, F. Circulating Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Cardiac Amyloidosis. J. Am. Heart Assoc. 2013, 2, e005868. [Google Scholar] [CrossRef] [Green Version]
- Slamova, I.; Adib, R.; Ellmerich, S.; Golos, M.R.; Gilbertson, J.A.; Botcher, N.; Canetti, D.; Taylor, G.W.; Rendell, N.; Tennent, G.A.; et al. Plasmin Activity Promotes Amyloid Deposition in a Transgenic Model of Human Transthyretin Amyloidosis. Nat. Commun. 2021, 12, 7112. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The Plasmin System Is Induced by and Degrades Amyloid-Beta Aggregates. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef] [Green Version]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM Structure of Cardiac Amyloid Fibrils from an Immunoglobulin Light Chain AL Amyloidosis Patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [Green Version]
- Radamaker, L.; Lin, Y.-H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM Structure of a Light Chain-Derived Amyloid Fibril from a Patient with Systemic AL Amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [Green Version]
- Radamaker, L.; Karimi-Farsijani, S.; Andreotti, G.; Baur, J.; Neumann, M.; Schreiner, S.; Berghaus, N.; Motika, R.; Haupt, C.; Walther, P.; et al. Role of Mutations and Post-Translational Modifications in Systemic AL Amyloidosis Studied by Cryo-EM. Nat. Commun. 2021, 12, 6434. [Google Scholar] [CrossRef]
- Tennent, G.A.; Lovat, L.B.; Pepys, M.B. Serum Amyloid P Component Prevents Proteolysis of the Amyloid Fibrils of Alzheimer Disease and Systemic Amyloidosis. Proc. Natl. Acad. Sci. USA 1995, 92, 4299–4303. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.J.; Ramirez-Alvarado, M. Glycosaminoglycans Promote Fibril Formation by Amyloidogenic Immunoglobulin Light Chains through a Transient Interaction. Biophys. Chem. 2011, 158, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Blancas-Mejía, L.M.; Hammernik, J.; Marin-Argany, M.; Ramirez-Alvarado, M. Differential Effects on Light Chain Amyloid Formation Depend on Mutations and Type of Glycosaminoglycans. J. Biol. Chem. 2015, 290, 4953–4965. [Google Scholar] [CrossRef] [Green Version]
- Greene, M.J.; Sam, F.; Soo Hoo, P.T.; Patel, R.S.; Seldin, D.C.; Connors, L.H. Evidence for a Functional Role of the Molecular Chaperone Clusterin in Amyloidotic Cardiomyopathy. Am. J. Pathol. 2011, 178, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Zhang, P.; Zhu, M.; Fabbri, E.; Gonzalez-Freire, M.; Moaddel, R.; Geng-Spyropoulos, M.; Ferrucci, L. A Targeted Proteomic Assay for the Measurement of Plasma Proteoforms Related to Human Aging Phenotypes. Proteomics 2017, 17, 1600232. [Google Scholar] [CrossRef]
- Berthelot, K.; Cullin, C.; Lecomte, S. What Does Make an Amyloid Toxic: Morphology, Structure or Interaction with Membrane? Biochimie 2013, 95, 12–19. [Google Scholar] [CrossRef]
- Cecchi, C.; Stefani, M. The Amyloid-Cell Membrane System. The Interplay between the Biophysical Features of Oligomers/Fibrils and Cell Membrane Defines Amyloid Toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef]
- Merlini, G.; Lousada, I.; Ando, Y.; Dispenzieri, A.; Gertz, M.A.; Grogan, M.; Maurer, M.S.; Sanchorawala, V.; Wechalekar, A.; Palladini, G.; et al. Rationale, Application and Clinical Qualification for NT-ProBNP as a Surrogate End Point in Pivotal Clinical Trials in Patients with AL Amyloidosis. Leukemia 2016, 30, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Lavatelli, F.; Russo, P.; Perlini, S.; Perfetti, V.; Bosoni, T.; Obici, L.; Bradwell, A.R.; D’Eril, G.M.; Fogari, R.; et al. Circulating Amyloidogenic Free Light Chains and Serum N-Terminal Natriuretic Peptide Type B Decrease Simultaneously in Association with Improvement of Survival in AL. Blood 2006, 107, 3854–3858. [Google Scholar] [CrossRef]
- Diomede, L.; Rognoni, P.; Lavatelli, F.; Romeo, M.; del Favero, E.; Cantù, L.; Ghibaudi, E.; di Fonzo, A.; Corbelli, A.; Fiordaliso, F.; et al. A Caenorhabditis Elegans–Based Assay Recognizes Immunoglobulin Light Chains Causing Heart Amyloidosis. Blood 2014, 123, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Diomede, L.; Romeo, M.; Rognoni, P.; Beeg, M.; Foray, C.; Ghibaudi, E.; Palladini, G.; Cherny, R.A.; Verga, L.; Capello, G.L.; et al. Cardiac Light Chain Amyloidosis: The Role of Metal Ions in Oxidative Stress and Mitochondrial Damage. Antioxid. Redox Signal. 2017, 27, 567–582. [Google Scholar] [CrossRef]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; del Monte, F.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; et al. Amyloidogenic Light Chains Induce Cardiomyocyte Contractile Dysfunction and Apoptosis via a Non-Canonical P38alpha MAPK Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Mishra, S.; Qiu, Y.; Shi, J.; Trudeau, K.; Las, G.; Liesa, M.; Shirihai, O.S.; Connors, L.H.; Seldin, D.C.; et al. Lysosomal Dysfunction and Impaired Autophagy Underlie the Pathogenesis of Amyloidogenic Light Chain-Mediated Cardiotoxicity. EMBO Mol. Med. 2014, 6, 1493–1507. [Google Scholar] [CrossRef]
- Brenner, D.A.; Jain, M.; Pimentel, D.R.; Wang, B.; Connors, L.H.; Skinner, M.; Apstein, C.S.; Liao, R. Human Amyloidogenic Light Chains Directly Impair Cardiomyocyte Function through an Increase in Cellular Oxidant Stress. Circ. Res. J. Am. Heart Assoc. 2004, 94, 1008–1010. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Joshi, S.; Ward, J.E.; Buys, E.P.; Mishra, D.; Mishra, D.; Morgado, I.; Fisch, S.; Lavatelli, F.; Merlini, G.; et al. Zebrafish Model of Amyloid Light Chain Cardiotoxicity: Regeneration versus Degeneration. Am. J. Physiol. 2019, 316, H1158–H1166. [Google Scholar] [CrossRef]
- Martin, E.B.; Williams, A.D.; Heidel, R.E.; Foster, J.S.; Lands, R.H.; Kennel, S.J.; Wall, J.S. A Functional Assay to Identify Amyloidogenic Light Chains. Amyloid 2018, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Zeldenrust, S.; Leung, N.; Kyle, R.A.; Russell, S.; et al. Coexistent Multiple Myeloma or Increased Bone Marrow Plasma Cells Define Equally High-Risk Populations in Patients with Immunoglobulin Light Chain Amyloidosis. J. Clin. Oncol. 2013, 31, 4319–4324. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Kyle, R.A.; Lacy, M.Q.; Therneau, T.M.; Larson, D.R.; Plevak, M.F.; Rajkumar, S.V.; Fonseca, R.; Greipp, P.R.; Witzig, T.E.; et al. Superior Survival in Primary Systemic Amyloidosis Patients Undergoing Peripheral Blood Stem Cell Transplantation: A Case-Control Study. Blood J. Hematol. 2004, 103, 3960–3963. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Dispenzieri, A.; Gertz, M.A. Evolution of Hematopoietic Cell Transplantation for Immunoglobulin Light Chain Amyloidosis. Biol. Blood Marrow Transplant. 2016, 22, 796–801. [Google Scholar] [CrossRef]
- Palladini, G.; Perfetti, V.; Obici, L.; Caccialanza, R.; Semino, A.; Adami, F.; Cavallero, G.; Rustichelli, R.; Virga, G.; Merlini, G. Association of Melphalan and High-Dose Dexamethasone Is Effective and Well Tolerated in Patients with AL (Primary) Amyloidosis Who Are Ineligible for Stem Cell Transplantation. Blood J. Hematol. 2004, 103, 2936–2938. [Google Scholar] [CrossRef]
- Shimazaki, C.; Fuchida, S.-I.; Suzuki, K.; Ishida, T.; Imai, H.; Sawamura, M.; Takamatsu, H.; Abe, M.; Miyamoto, T.; Hata, H.; et al. Phase 1 Study of Bortezomib in Combination with Melphalan and Dexamethasone in Japanese Patients with Relapsed AL Amyloidosis. Int. J. Hematol. 2016, 103, 79–85. [Google Scholar] [CrossRef]
- Shimazaki, C.; Hata, H.; Iida, S.; Ueda, M.; Katoh, N.; Sekijima, Y.; Ikeda, S.; Yazaki, M.; Fukushima, W.; Ando, Y. Nationwide Survey of 741 Patients with Systemic Amyloid Light-Chain Amyloidosis in Japan. Intern. Med. 2018, 57, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and Its Role in the Management of Patients with Multiple Myeloma. Expert Rev. Anticancer Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jia, Y.; Song, W.; Zhang, L. Therapeutic Potential of Nitrogen Mustard Based Hybrid Molecules. Front. Pharmacol. 2018, 9, 1453. [Google Scholar] [CrossRef]
- Ralhan, R.; Kaur, J. Alkylating Agents and Cancer Therapy. Expert Opin. Ther. Pat. 2007, 17, 1061–1075. [Google Scholar] [CrossRef]
- Kyle, R.A.; Greipp, P.R. Primary Systemic Amyloidosis: Comparison of Melphalan and Prednisone versus Placebo. Blood J. Hematol. 1978, 52, 818–827. [Google Scholar]
- Palladini, G.; Russo, P.; Nuvolone, M.; Lavatelli, F.; Perfetti, V.; Obici, L.; Merlini, G. Treatment with Oral Melphalan plus Dexamethasone Produces Long-Term Remissions in AL Amyloidosis. Blood J. Hematol. 2007, 110, 787–788. [Google Scholar] [CrossRef] [Green Version]
- Sanchorawala, V.; Seldin, D.C.; Berk, J.L.; Sloan, J.M.; Doros, G.; Skinner, M. Oral Cyclic Melphalan and Dexamethasone for Patients with AL Amyloidosis. Clin. Lymphoma Myeloma Leuk. 2010, 10, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Hussein, M.A.; Rasmussen, E.; Solomon, A.; Larson, R.A.; Crowley, J.J.; Barlogie, B. Clinical Efficacy of High-Dose Dexamethasone with Maintenance Dexamethasone/Alpha Interferon in Patients with Primary Systemic Amyloidosis: Results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood J. Hematol. 2004, 104, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and Cancer: Golden Anniversary. Nat. Rev. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- West, N.J. Prevention and Treatment of Hemorrhagic Cystitis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1997, 17, 696–706. [Google Scholar] [CrossRef]
- Talar-Williams, C.; Hijazi, Y.M.; Walther, M.M.; Linehan, W.M.; Hallahan, C.W.; Lubensky, I.; Kerr, G.S.; Hoffman, G.S.; Fauci, A.S.; Sneller, M.C. Cyclophosphamide-Induced Cystitis and Bladder Cancer in Patients with Wegener Granulomatosis. Ann. Intern. Med. 1996, 124, 477–484. [Google Scholar] [CrossRef]
- Bianchi, G.; Ghobrial, I.M. Molecular Mechanisms of Effectiveness of Novel Therapies in Multiple Myeloma. Leuk. Lymphoma 2013, 54, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Briemberg, H.; Jagannath, S.; Wen, P.Y.; Barlogie, B.; Berenson, J.; Singhal, S.; Siegel, D.S.; Irwin, D.; Schuster, M.; et al. Frequency, Characteristics, and Reversibility of Peripheral Neuropathy during Treatment of Advanced Multiple Myeloma with Bortezomib. J. Clin. Oncol. 2006, 24, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Voortman, J.; Giaccone, G. Severe Reversible Cardiac Failure after Bortezomib Treatment Combined with Chemotherapy in a Non-Small Cell Lung Cancer Patient: A Case Report. BMC Cancer 2006, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Hacihanefioglu, A.; Tarkun, P.; Gonullu, E. Acute Severe Cardiac Failure in a Myeloma Patient Due to Proteasome Inhibitor Bortezomib. Int. J. Hematol. 2008, 88, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, M.; Chakravarty, S.; Schulman, P.; Bockorny, B.; Bona, R. Severe Heart Failure after Bortezomib Treatment in a Patient with Multiple Myeloma: A Case Report and Review of the Literature. Acta Haematol. 2012, 128, 244–247. [Google Scholar] [CrossRef]
- Enrico, O.; Gabriele, B.; Nadia, C.; Sara, G.; Daniele, V.; Giulia, C.; Antonio, S.; Mario, P. Unexpected Cardiotoxicity in Haematological Bortezomib Treated Patients. Br. J. Haematol. 2007, 138, 396–397. [Google Scholar] [CrossRef] [PubMed]
- Nowis, D.; Maczewski, M.; Mackiewicz, U.; Kujawa, M.; Ratajska, A.; Wieckowski, M.R.; Wilczyński, G.M.; Malinowska, M.; Bil, J.; Salwa, P.; et al. Cardiotoxicity of the Anticancer Therapeutic Agent Bortezomib. Am. J. Pathol. 2010, 176, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Wohlert, C.; Saguner, A.M.; Flores, A.; Nesbitt, L.L.; Chade, A.; Lerman, L.O.; Lerman, A. Primary Proteasome Inhibition Results in Cardiac Dysfunction. Eur. J. Heart Fail. 2013, 15, 614–623. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Sachchithanantham, S.; Milani, P.; Gillmore, J.; Foli, A.; Lachmann, H.; Basset, M.; Hawkins, P.; Merlini, G.; Wechalekar, A.D. A European Collaborative Study of Cyclophosphamide, Bortezomib, and Dexamethasone in Upfront Treatment of Systemic AL Amyloidosis. Blood J. Am. Soc. Hematol. 2015, 126, 612–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J. The Development of Proteasome Inhibitors as Anticancer Drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma Cell Differentiation Requires the Transcription Factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-[Kappa]B Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-KappaB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib Induces Canonical Nuclear Factor-KappaB Activation in Multiple Myeloma Cells. Blood J. Am. Soc. Hematol. 2009, 114, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.W.C.J.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W.H.I. Monoclonal Antibodies Targeting CD38 in Hematological Malignancies and Beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Van de Wyngaert, Z.; Carpentier, B.; Pascal, L.; Lionne-Huyghe, P.; Leduc, I.; Srour, M.; Vasseur, M.; Demarquette, H.; Terriou, L.; Herbaux, C.; et al. Daratumumab Is Effective in the Relapsed or Refractory Systemic Light-Chain Amyloidosis but Associated with High Infection Burden in a Frail Real-Life Population. Br. J. Haematol. 2020, 188, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Sanchorawala, V.; Sarosiek, S.; Schulman, A.; Mistark, M.; Migre, M.E.; Cruz, R.; Sloan, J.M.; Brauneis, D.; Shelton, A.C. Safety, Tolerability, and Response Rates of Daratumumab in Relapsed AL Amyloidosis: Results of a Phase 2 Study. Blood J. Am. Soc. Hematol. 2020, 135, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Fenton, B.; Akhtar, A.; Gertz, M.A. First Report of Safety and Efficacy of Daratumumab in 2 Cases of Advanced Immunoglobulin Light Chain Amyloidosis. Blood J. Am. Soc. Hematol. 2016, 128, 1987–1989. [Google Scholar] [CrossRef] [Green Version]
- Roussel, M.; Merlini, G.; Chevret, S.; Arnulf, B.; Stoppa, A.M.; Perrot, A.; Palladini, G.; Karlin, L.; Royer, B.; Huart, A.; et al. A Prospective Phase 2 Trial of Daratumumab in Patients with Previously Treated Systemic Light-Chain Amyloidosis. Blood J. Am. Soc. Hematol. 2020, 135, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Vrana, J.A.; Theis, J.D.; Dasari, S.; Mereuta, O.M.; Dispenzieri, A.; Zeldenrust, S.R.; Gertz, M.A.; Kurtin, P.J.; Grogg, K.L.; Dogan, A. Clinical Diagnosis and Typing of Systemic Amyloidosis in Subcutaneous Fat Aspirates by Mass Spectrometry-Based Proteomics. Haematologica 2014, 99, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Warsame, R.; LaPlant, B.; Kumar, S.K.; Laumann, K.; Perez Burbano, G.; Buadi, F.K.; Gertz, M.A.; Kyle, R.A.; Lacy, M.Q.; Dingli, D.; et al. Long-Term Outcomes of IMiD-Based Trials in Patients with Immunoglobulin Light-Chain Amyloidosis: A Pooled Analysis. Blood Cancer J. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.V.; Bhutani, D.; Mapara, M.; Radhakrishnan, J.; Shames, S.; Maurer, M.S.; Leng, S.; Wall, J.S.; Solomon, A.; Eisenberger, A.; et al. One Year Follow up Analysis of the Phase 1a/b Study of Chimeric Fibril-Reactive Monoclonal Antibody 11-1F4 in Patients with AL Amyloidosis. Amyloid 2019, 26, 115–116. [Google Scholar] [CrossRef]
- Edwards, C.V.; Rao, N.; Bhutani, D.; Mapara, M.; Radhakrishnan, J.; Shames, S.; Maurer, M.S.; Leng, S.; Solomon, A.; Lentzsch, S.; et al. Phase 1a/b Study of Monoclonal Antibody CAEL-101 (11-1F4) in Patients with AL Amyloidosis. Blood 2021, 138, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Gertz, M.A.; Kyle, R.A.; Lacy, M.Q.; Burritt, M.F.; Therneau, T.M.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Rajkumar, S.V.; et al. Serum Cardiac Troponins and N-Terminal pro-Brain Natriuretic Peptide: A Staging System for Primary Systemic Amyloidosis. J. Clin. Oncol. 2004, 22, 3751–3757. [Google Scholar] [CrossRef]
- Goto, S.; Mahara, K.; Beussink-Nelson, L.; Ikura, H.; Katsumata, Y.; Endo, J.; Gaggin, H.K.; Shah, S.J.; Itabashi, Y.; MacRae, C.A.; et al. Artificial Intelligence-Enabled Fully Automated Detection of Cardiac Amyloidosis Using Electrocardiograms and Echocardiograms. Nat. Commun. 2021, 12, 2726. [Google Scholar] [CrossRef]
- Brons, M.; Muller, S.A.; Rutten, F.H.; van der Meer, M.G.; Vrancken, A.F.J.E.; Minnema, M.C.; Baas, A.F.; Asselbergs, F.W.; Oerlemans, M.I.F.J. Evaluation of the Cardiac Amyloidosis Clinical Pathway Implementation: A Real-World Experience. Eur. Heart J. Open 2022, 2, oeac011. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikura, H.; Endo, J.; Kitakata, H.; Moriyama, H.; Sano, M.; Fukuda, K. Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis. Int. J. Mol. Sci. 2022, 23, 6336. https://doi.org/10.3390/ijms23116336
Ikura H, Endo J, Kitakata H, Moriyama H, Sano M, Fukuda K. Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis. International Journal of Molecular Sciences. 2022; 23(11):6336. https://doi.org/10.3390/ijms23116336
Chicago/Turabian StyleIkura, Hidehiko, Jin Endo, Hiroki Kitakata, Hidenori Moriyama, Motoaki Sano, and Keiichi Fukuda. 2022. "Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis" International Journal of Molecular Sciences 23, no. 11: 6336. https://doi.org/10.3390/ijms23116336
APA StyleIkura, H., Endo, J., Kitakata, H., Moriyama, H., Sano, M., & Fukuda, K. (2022). Molecular Mechanism of Pathogenesis and Treatment Strategies for AL Amyloidosis. International Journal of Molecular Sciences, 23(11), 6336. https://doi.org/10.3390/ijms23116336