
Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales


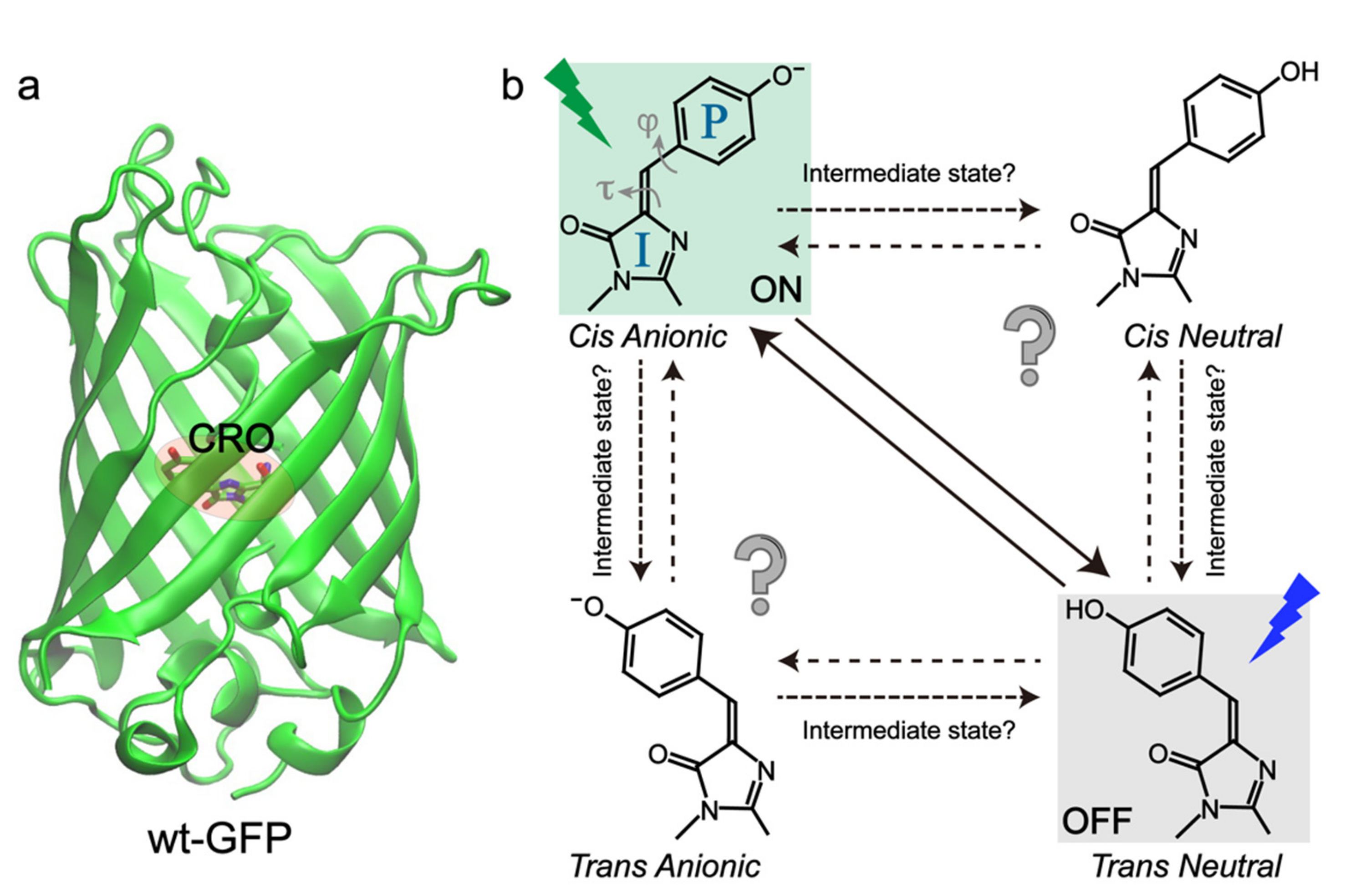{kind=link}
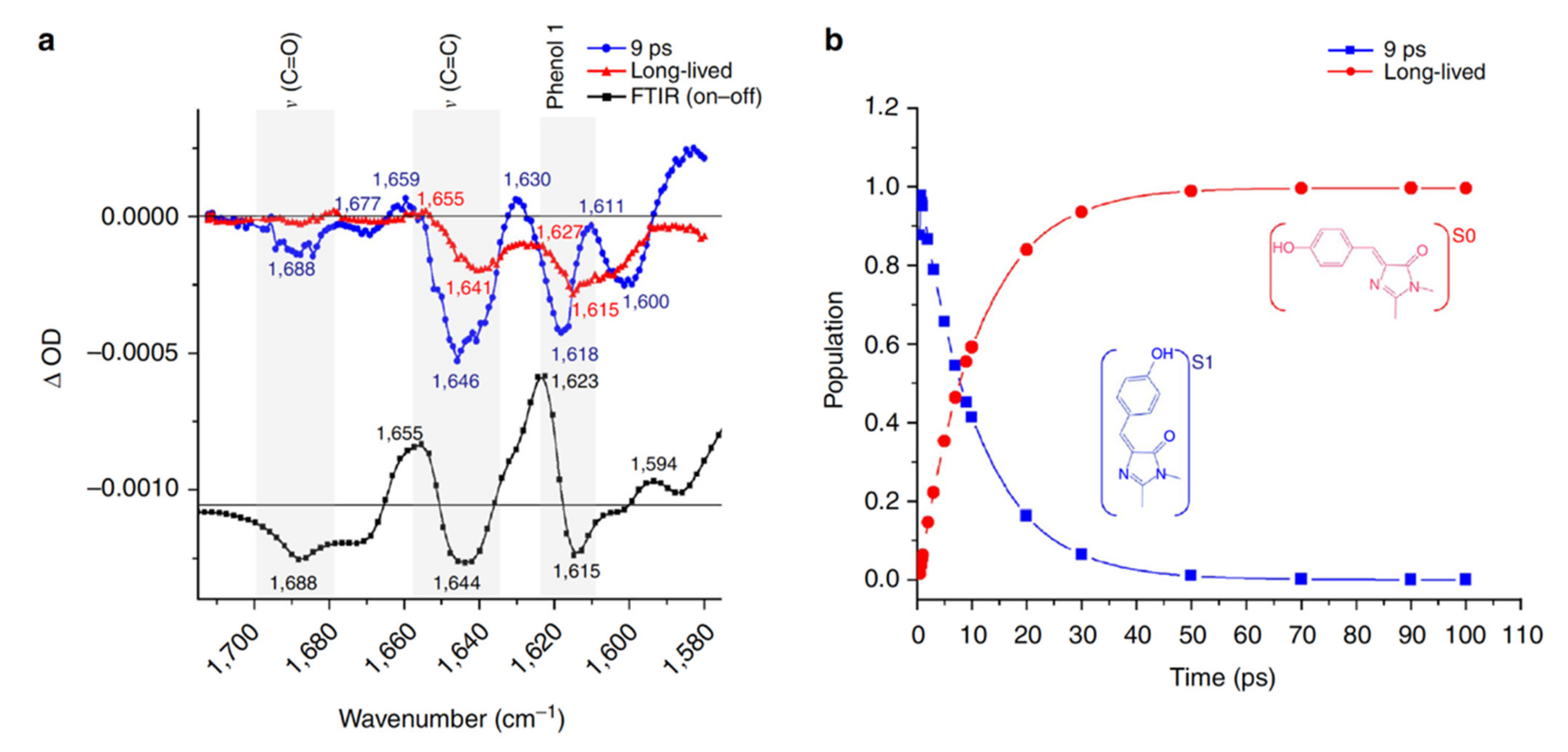{kind=link}
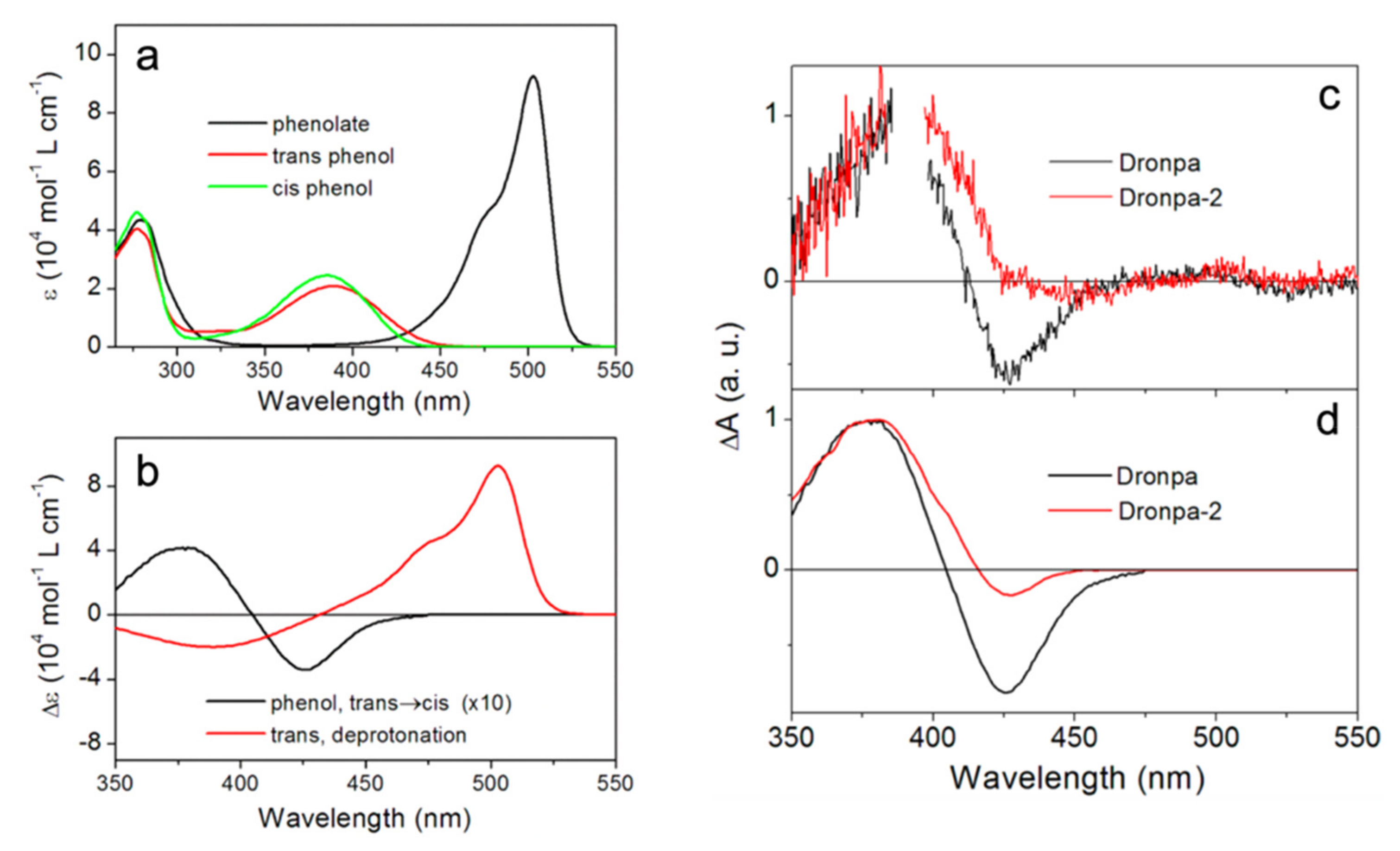{kind=link}
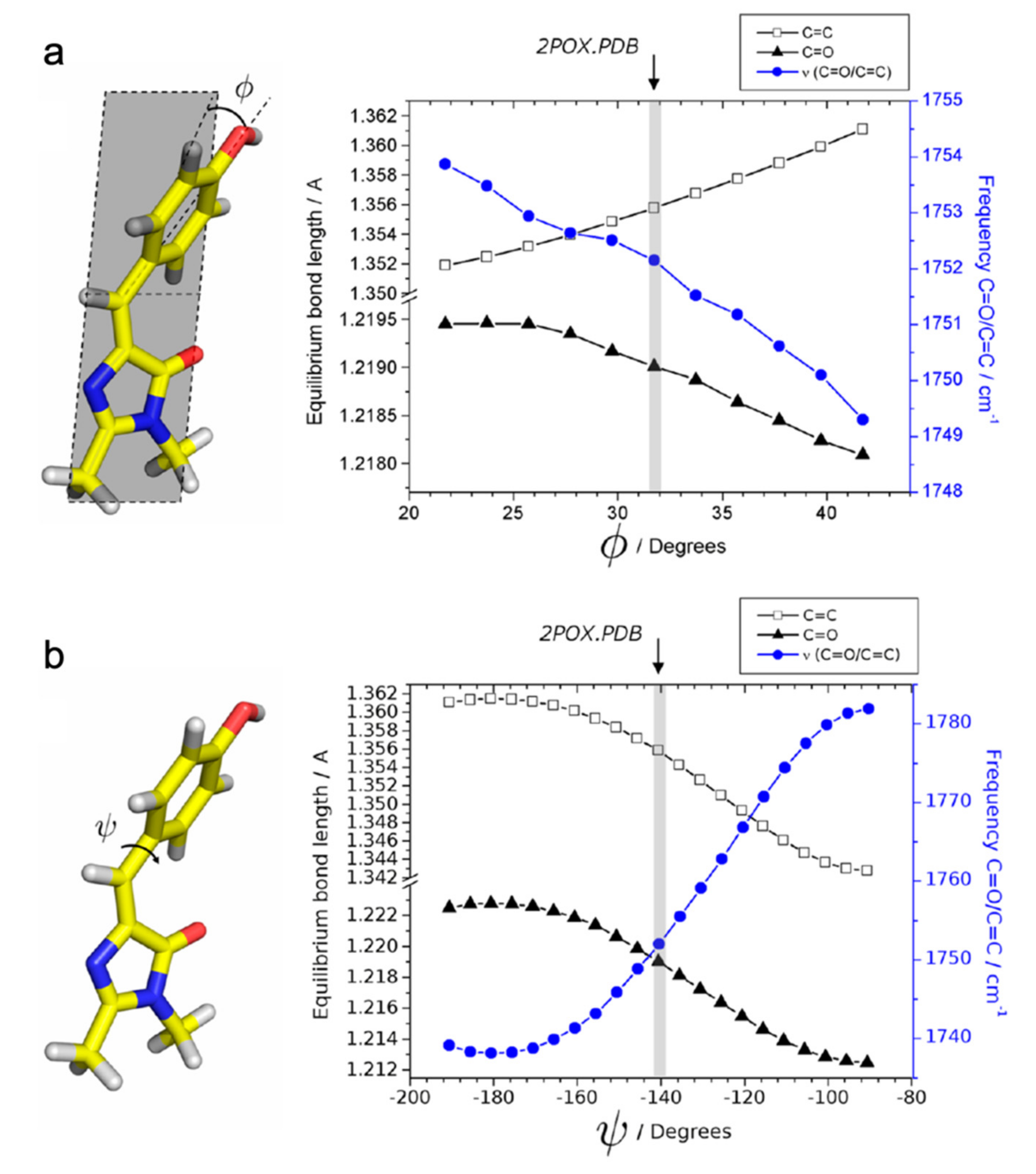{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Negative RSFPs
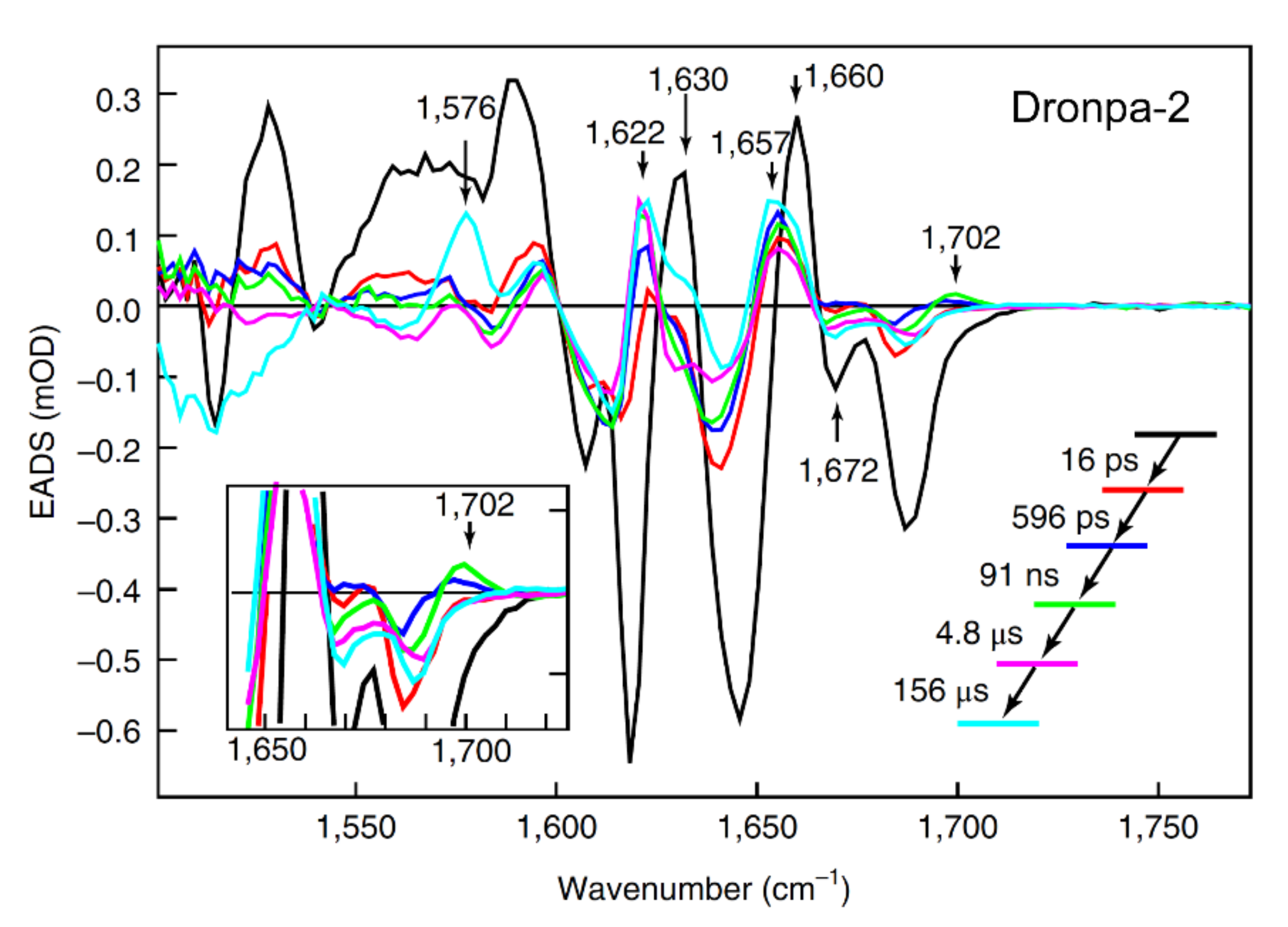
2.1. Dronpa and Dronpa-2
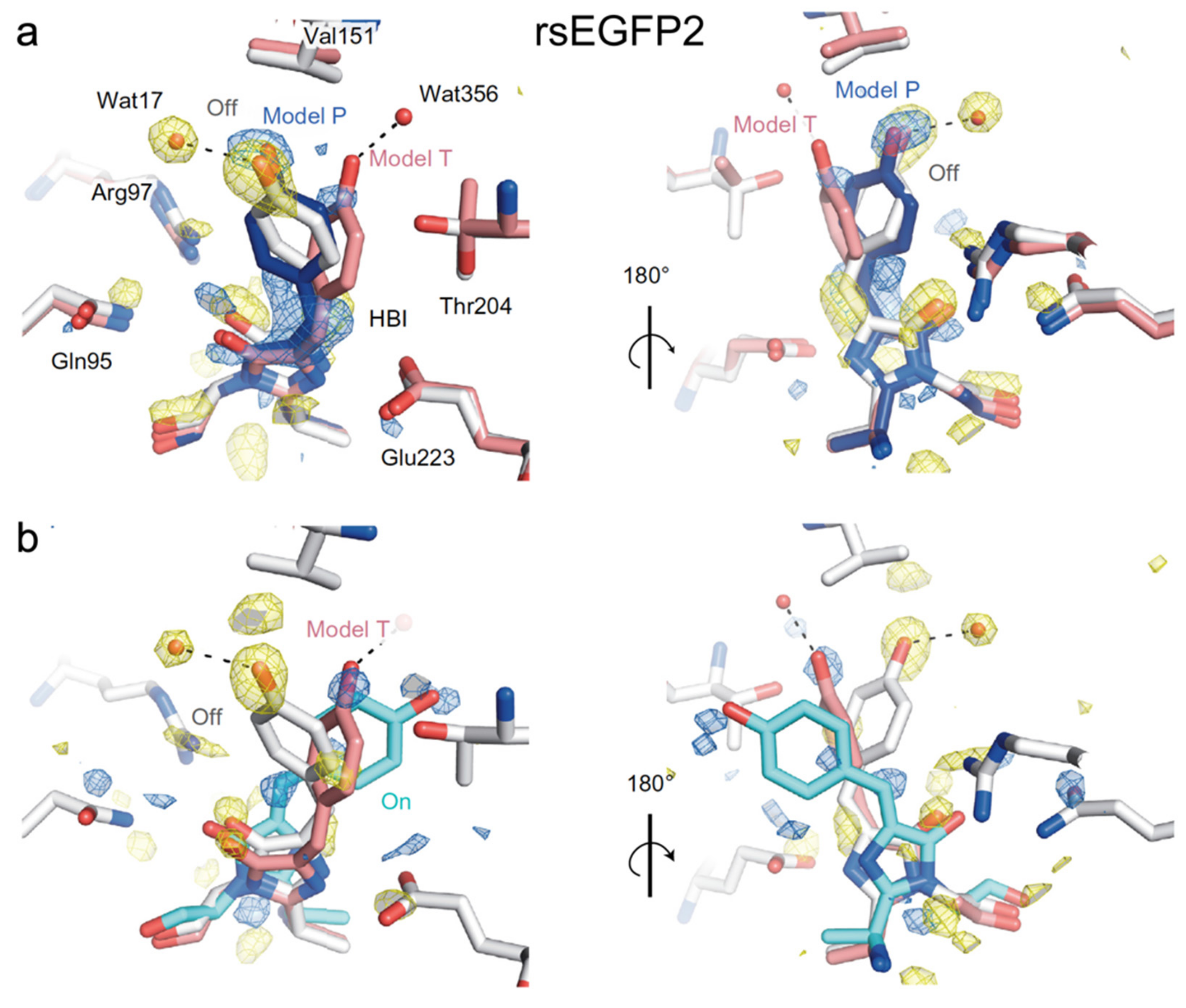
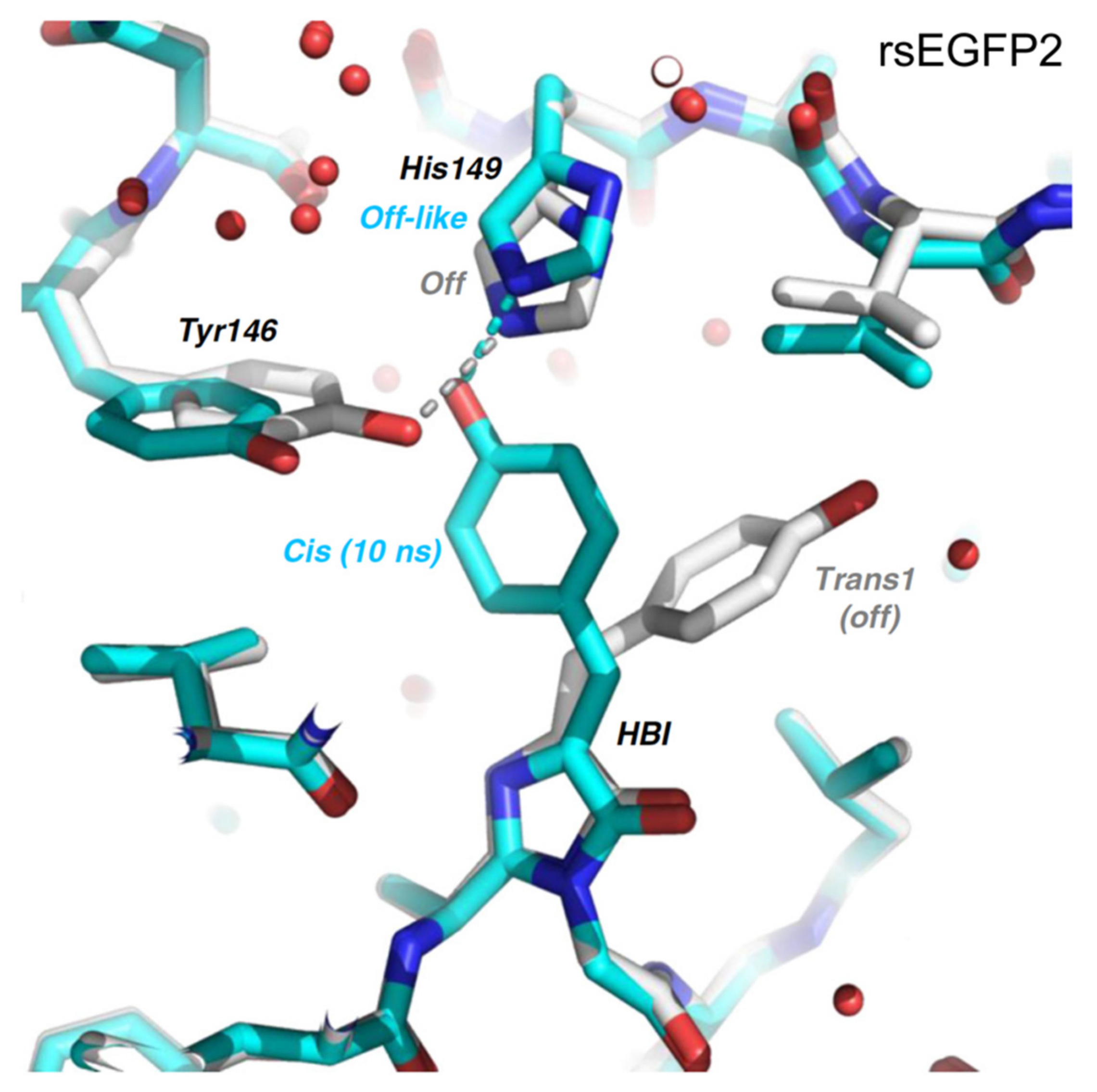
2.2. RsEGFP2
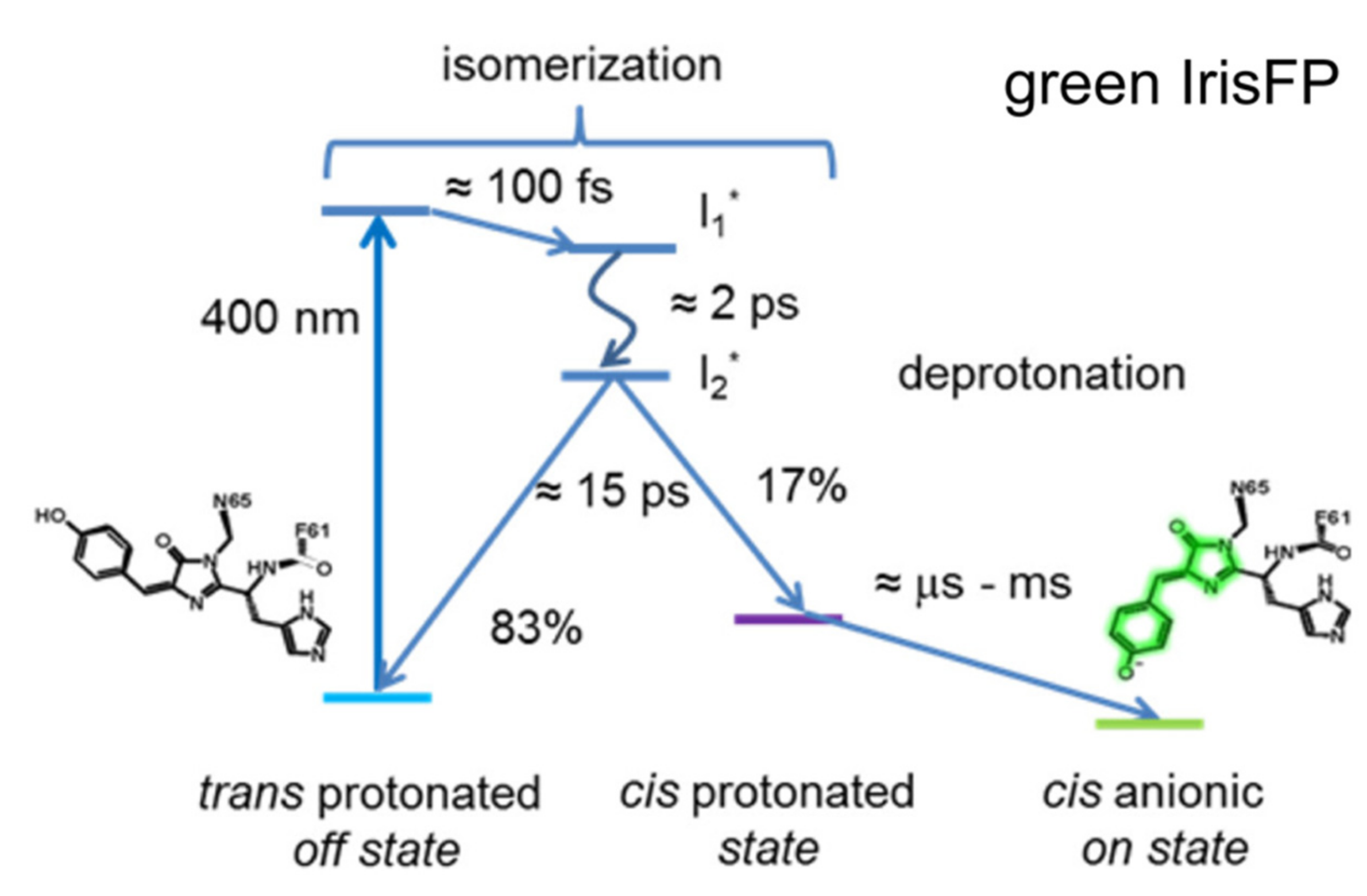
2.3. IrisFP
2.4. Other Negative RSFPs
3. Positive RSFPs
4. Decoupled RSFPs
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–781. [Google Scholar] [CrossRef]
- Jung, G. (Ed.) Fluorescent Proteins II: Application of Fluorescent Protein Technology; Springer: Berlin/Heidelberg, Germany, 2012; p. 284. [Google Scholar]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moerner, W.E. New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12596–12602. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction barrier: Super-resolution imaging of cells. Cell 2010, 143, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotjohann, T.; Testa, I.; Leutenegger, M.; Bock, H.; Urban, N.T.; Lavoie-Cardinal, F.; Willig, K.I.; Eggeling, C.; Jakobs, S.; Hell, S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 2011, 478, 204–208. [Google Scholar] [CrossRef]
- Chozinski, T.J.; Gagnon, L.A.; Vaughan, J.C. Twinkle, twinkle little star: Photoswitchable fluorophores for super-resolution imaging. FEBS Lett. 2014, 588, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakova, D.M.; Sengupta, P.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photocontrollable fluorescent proteins for superresolution imaging. Annu. Rev. Biophys. 2014, 43, 303–329. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491. [Google Scholar] [CrossRef]
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Wachter, M.R. Photoconvertible fluorescent proteins and the role of dynamics in protein evolution. Int. J. Mol. Sci. 2017, 18, 1792. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, K.A.; Fradkov, A.F.; Gurskaya, N.G.; Matz, M.V.; Labas, Y.A.; Savitsky, A.P.; Markelov, M.L.; Zaraisky, A.G.; Zhao, X.; Fang, Y.; et al. Natural animal coloration can be determined by a nonfluorescent green fluorescent protein homolog. J. Biol. Chem. 2000, 275, 25879–25882. [Google Scholar] [CrossRef] [Green Version]
- Piatkevich, K.D.; English, B.P.; Malashkevich, V.N.; Xiao, H.; Almo, S.C.; Singer, R.H.; Verkhusha, V.V. Photoswitchable red fluorescent protein with a large Stokes shift. Chem. Biol. 2014, 21, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, M.; Li, D.; He, W.; Peng, J.; Betzig, E.; Xu, P. Highly photostable, reversibly photoswitchable fluorescent protein with high contrast ratio for live-cell superresolution microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, 10364–10369. [Google Scholar] [CrossRef] [Green Version]
- Pennacchietti, F.; Serebrovskaya, E.O.; Faro, A.R.; Shemyakina, I.I.; Bozhanova, N.G.; Kotlobay, A.A.; Gurskaya, N.G.; Bodén, A.; Dreier, J.; Chudakov, D.M.; et al. Fast reversibly photoswitching red fluorescent proteins for live-cell RESOLFT nanoscopy. Nat. Methods 2018, 15, 601–604. [Google Scholar] [CrossRef]
- Wazawa, T.; Noma, R.; Uto, S.; Sugiura, K.; Washio, T.; Nagai, T. A photoswitchable fluorescent protein for hours-time-lapse and sub-second-resolved super-resolution imaging. Microscopy 2021, 70, 340–352. [Google Scholar] [CrossRef]
- Jensen, N.A.; Jansen, I.; Kamper, M.; Jakobs, S. Reversibly switchable fluorescent proteins for RESOLFT nanoscopy. In Nanoscale Photonic Imaging; Salditt, T., Egner, A., Luke, D.R., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 241–261. [Google Scholar]
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373. [Google Scholar] [CrossRef]
- Andresen, M.; Stiel, A.C.; Fölling, J.; Wenzel, D.; Schönle, A.; Egner, A.; Eggeling, C.; Hell, S.W.; Jakobs, S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008, 26, 1035–1040. [Google Scholar] [CrossRef]
- Brakemann, T.; Stiel, A.C.; Weber, G.; Andresen, M.; Testa, I.; Grotjohann, T.; Leutenegger, M.; Plessmann, U.; Urlaub, H.; Eggeling, C.; et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat. Biotechnol. 2011, 29, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Takauchi, H.; Ogami, Y.; Fujiwara, S.; Nakano, M.; Matsuda, T.; Nagai, T. Spontaneously blinking fluorescent protein for simple single laser super-resolution live cell imaging. ACS Chem. Biol. 2018, 13, 1938–1943. [Google Scholar] [CrossRef] [PubMed]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remington, S.J. Green fluorescent protein: A perspective. Protein Sci. 2011, 20, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Jung, G. (Ed.) Fluorescent Proteins I: From Understanding to Design; Springer: Berlin/Heidelberg, Germany, 2012; Volume 11, p. 268. [Google Scholar]
- Fanjiang, M.-W.; Li, M.-J.; Sung, R.; Sung, K. Synthesis and properties of the para-trimethylammonium analogues of green fluorescence protein (GFP) chromophore: The mimic of protonated GFP chromophore. Bioorg. Chem. 2018, 77, 300–310. [Google Scholar] [CrossRef]
- Andresen, M.; Wahl, M.C.; Stiel, A.C.; Gräter, F.; Schäfer, L.V.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13070–13074. [Google Scholar] [CrossRef] [Green Version]
- Quillin, M.L.; Anstrom, D.M.; Shu, X.; O’Leary, S.; Kallio, K.; Chudakov, D.M.; Remington, S.J. Kindling fluorescent protein from Anemonia sulcata: Dark-state structure at 1.38 Å resolution. Biochemistry 2005, 44, 5774–5787. [Google Scholar] [CrossRef]
- Grigorenko, B.; Savitsky, A.; Topol, I.; Burt, S.; Nemukhin, A. Ground-state structures and vertical excitations for the kindling fluorescent protein asFP595. J. Phys. Chem. B 2006, 110, 18635–18640. [Google Scholar] [CrossRef]
- Andresen, M.; Stiel, A.C.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Wahl, M.C.; Hell, S.W.; Jakobs, S. Structural basis for reversible photoswitching in Dronpa. Proc. Natl. Acad. Sci. USA 2007, 104, 13005–13009. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.N.; Ai, H.-w.; Campbell, R.E.; Remington, S.J. Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proc. Natl. Acad. Sci. USA 2007, 104, 6672–6677. [Google Scholar] [CrossRef] [Green Version]
- Moors, S.L.C.; Michielssens, S.; Flors, C.; Dedecker, P.; Hofkens, J.; Ceulemans, A. How is cis-trans isomerization controlled in Dronpa mutants? A replica exchange molecular dynamics study. J. Chem. Theory Comput. 2008, 4, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Brakemann, T.; Weber, G.; Andresen, M.; Groenhof, G.; Stiel, A.C.; Trowitzsch, S.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; Wahl, M.C.; et al. Molecular basis of the light-driven switching of the photochromic fluorescent protein Padron. J. Biol. Chem. 2010, 285, 14603–14609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chung, L.W.; Mizuno, H.; Miyawaki, A.; Morokuma, K. Primary events of photodynamics in reversible photoswitching fluorescent protein Dronpa. J. Phys. Chem. Lett. 2010, 1, 3328–3333. [Google Scholar] [CrossRef]
- Acharya, A.; Bogdanov, A.M.; Grigorenko, B.L.; Bravaya, K.B.; Nemukhin, A.V.; Lukyanov, K.A.; Krylov, A.I. Photoinduced chemistry in fluorescent proteins: Curse or blessing? Chem. Rev. 2016, 117, 758–795. [Google Scholar] [CrossRef]
- Gozem, S.; Luk, H.L.; Schapiro, I.; Olivucci, M. Theory and simulation of the ultrafast double-bond isomerization of biological chromophores. Chem. Rev. 2017, 117, 13502–13565. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Tang, L.; Chen, C. Unveiling coupled electronic and vibrational motions of chromophores in condensed phases. J. Chem. Phys. 2019, 151, 200901. [Google Scholar] [CrossRef]
- Romei, M.G.; Lin, C.-Y.; Mathews, I.I.; Boxer, S.G. Electrostatic control of photoisomerization pathways in proteins. Science 2020, 367, 76–79. [Google Scholar] [CrossRef]
- Jones, C.M.; List, N.H.; Martínez, T.J. Resolving the ultrafast dynamics of the anionic green fluorescent protein chromophore in water. Chem. Sci. 2021, 12, 11347–11363. [Google Scholar] [CrossRef]
- Tang, L.; Fang, C. Fluorescence modulation by ultrafast chromophore twisting events: Developing a powerful toolset for fluorescent-protein-based imaging. J. Phys. Chem. B 2021, 125, 13610–13623. [Google Scholar] [CrossRef]
- Mukherjee, S.; Jimenez, R. Photophysical engineering of fluorescent proteins: Accomplishments and challenges of physical chemistry strategies. J. Phys. Chem. B 2022, 126, 735–750. [Google Scholar] [CrossRef]
- Fron, E.; Flors, C.; Schweitzer, G.; Habuchi, S.; Mizuno, H.; Ando, R.; De Schryver, F.C.; Miyawaki, A.; Hofkens, J. Ultrafast excited-state dynamics of the photoswitchable protein Dronpa. J. Am. Chem. Soc. 2007, 129, 4870–4871. [Google Scholar] [CrossRef] [PubMed]
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem. 2018, 10, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Laptenok, S.P.; Gil, A.A.; Hall, C.R.; Lukacs, A.; Iuliano, J.N.; Jones, G.A.; Greetham, G.M.; Donaldson, P.; Miyawaki, A.; Tonge, P.J.; et al. Infrared spectroscopy reveals multi-step multi-timescale photoactivation in the photoconvertible protein archetype Dronpa. Nat. Chem. 2018, 10, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Wilmann, P.G.; Turcic, K.; Battad, J.M.; Wilce, M.C.J.; Devenish, R.J.; Prescott, M.; Rossjohn, J. The 1.7 Å crystal structure of Dronpa: A photoswitchable green fluorescent protein. J. Mol. Biol. 2006, 364, 213–224. [Google Scholar] [CrossRef]
- Stiel, A.C.; Trowitzsch, S.; Weber, G.; Andresen, M.; Eggeling, C.; Hell, S.W.; Jakobs, S.; Wahl, M.C. 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochem. J. 2007, 402, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, H.; Mal, T.K.; Walchli, M.; Kikuchi, A.; Fukano, T.; Ando, R.; Jeyakanthan, J.; Taka, J.; Shiro, Y.; Ikura, M.; et al. Light-dependent regulation of structural flexibility in a photochromic fluorescent protein. Proc. Natl. Acad. Sci. USA 2008, 105, 9227–9232. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chung, L.W.; Mizuno, H.; Miyawaki, A.; Morokuma, K. A theoretical study on the nature of on- and off-states of reversibly photoswitching fluorescent protein Dronpa: Absorption, emission, protonation, and Raman. J. Phys. Chem. B 2010, 114, 1114–1126. [Google Scholar] [CrossRef]
- Smyrnova, D.; Zinovjev, K.; Tuñón, I.; Ceulemans, A. Thermal isomerization mechanism in Dronpa and its mutants. J. Phys. Chem. B 2016, 120, 12820–12825. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Jablonski, A.E.; Issaeva, I.; Bourassa, D.; Hsiang, J.-C.; Fahrni, C.J.; Dickson, R.M. Optically modulated photoswitchable fluorescent proteins yield improved biological imaging sensitivity. J. Am. Chem. Soc. 2015, 137, 12764–12767. [Google Scholar] [CrossRef] [Green Version]
- Ando, R.; Flors, C.; Mizuno, H.; Hofkens, J.; Miyawaki, A. Highlighted generation of fluorescence signals using simultaneous two-color irradiation on Dronpa mutants. Biophys. J. 2007, 92, L97–L99. [Google Scholar] [CrossRef] [Green Version]
- Habuchi, S.; Ando, R.; Dedecker, P.; Verheijen, W.; Mizuno, H.; Miyawaki, A.; Hofkens, J. Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proc. Natl. Acad. Sci. USA 2005, 102, 9511–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, M.M.; Kaucikas, M.; Fitzpatrick, A.; Champion, P.; Timothy Sage, J.; van Thor, J.J. Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa. Nat. Commun. 2013, 4, 1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacs, A.; Haigney, A.; Brust, R.; Addison, K.; Towrie, M.; Greetham, G.M.; Jones, G.A.; Miyawaki, A.; Tonge, P.J.; Meech, S.R. Protein photochromism observed by ultrafast vibrational spectroscopy. J. Phys. Chem. B 2013, 117, 11954–11959. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200–204. [Google Scholar] [CrossRef]
- Han, F.; Liu, W.; Fang, C. Excited-state proton transfer of photoexcited pyranine in water observed by femtosecond stimulated Raman spectroscopy. Chem. Phys. 2013, 422, 204–219. [Google Scholar] [CrossRef]
- Yadav, D.; Lacombat, F.; Dozova, N.; Rappaport, F.; Plaza, P.; Espagne, A. Real-time monitoring of chromophore isomerization and deprotonation during the photoactivation of the fluorescent protein Dronpa. J. Phys. Chem. B 2015, 119, 2404–2414. [Google Scholar] [CrossRef]
- Higashino, A.; Mizuno, M.; Mizutani, Y. Chromophore structure of photochromic fluorescent protein Dronpa: Acid–base equilibrium of two cis configurations. J. Phys. Chem. B 2016, 120, 3353–3359. [Google Scholar] [CrossRef]
- Luin, S.; Voliani, V.; Lanza, G.; Bizzarri, R.; Nifosì, R.; Amat, P.; Tozzini, V.; Serresi, M.; Beltram, F. Raman study of chromophore states in photochromic fluorescent proteins. J. Am. Chem. Soc. 2009, 131, 96–103. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L. Mapping structural dynamics of proteins with femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2020, 71, 239–265. [Google Scholar] [CrossRef] [Green Version]
- Kaucikas, M.; Tros, M.; van Thor, J.J. Photoisomerization and proton transfer in the forward and reverse photoswitching of the fast-switching M159T mutant of the Dronpa fluorescent protein. J. Phys. Chem. B 2015, 119, 2350–2362. [Google Scholar] [CrossRef]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef] [PubMed]
- McCamant, D.W.; Kukura, P.; Yoon, S.; Mathies, R.A. Femtosecond broadband stimulated Raman spectroscopy: Apparatus and methods. Rev. Sci. Instrum. 2004, 75, 4971–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of Aequorin, a bioluminescent protein from the luminous Hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Brejc, K.; Sixma, T.K.; Kitts, P.A.; Kain, S.R.; Tsien, R.Y.; Ormö, M.; Remington, S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2306–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Grotjohann, T.; Testa, I.; Reuss, M.; Brakemann, T.; Eggeling, C.; Hell, S.W.; Jakobs, S. RsEGFP2 enables fast RESOLFT nanoscopy of living cells. eLife 2012, 1, e00248. [Google Scholar] [CrossRef]
- El Khatib, M.; Martins, A.; Bourgeois, D.; Colletier, J.-P.; Adam, V. Rational design of ultrastable and reversibly photoswitchable fluorescent proteins for super-resolution imaging of the bacterial periplasm. Sci. Rep. 2016, 6, 18459. [Google Scholar] [CrossRef] [Green Version]
- Uriarte, L.M.; Vitale, R.; Niziński, S.; Hadjidemetriou, K.; Zala, N.; Lukacs, A.; Greetham, G.M.; Sazanovich, I.V.; Weik, M.; Ruckebusch, C.; et al. Structural information about the trans-to-cis isomerization mechanism of the photoswitchable fluorescent protein rsEGFP2 revealed by multiscale infrared transient absorption. J. Phys. Chem. Lett. 2022, 13, 1194–1202. [Google Scholar] [CrossRef]
- Chang, J.; Romei, M.G.; Boxer, S.G. Structural evidence of photoisomerization pathways in fluorescent proteins. J. Am. Chem. Soc. 2019, 141, 15504–15508. [Google Scholar] [CrossRef]
- Adam, V.; Hadjidemetriou, K.; Jensen, N.; Shoeman, R.L.; Woodhouse, J.; Aquila, A.; Banneville, A.-S.; Barends, T.R.M.; Bezchastnov, V.; Boutet, S.; et al. Rational control of structural off-state heterogeneity in a photoswitchable fluorescent protein provides switching contrast enhancement. bioRxiv 2021. [Google Scholar] [CrossRef]
- Woodhouse, J.; Nass Kovacs, G.; Coquelle, N.; Uriarte, L.M.; Adam, V.; Barends, T.R.M.; Byrdin, M.; de la Mora, E.; Bruce Doak, R.; Feliks, M.; et al. Photoswitching mechanism of a fluorescent protein revealed by time-resolved crystallography and transient absorption spectroscopy. Nat. Commun. 2020, 11, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.-D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, V.; Lelimousin, M.; Boehme, S.; Desfonds, G.; Nienhaus, K.; Field, M.J.; Wiedenmann, J.; McSweeney, S.; Nienhaus, G.U.; Bourgeois, D. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proc. Natl. Acad. Sci. USA 2008, 105, 18343–18348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colletier, J.-P.; Sliwa, M.; Gallat, F.-X.; Sugahara, M.; Guillon, V.; Schirò, G.; Coquelle, N.; Woodhouse, J.; Roux, L.; Gotthard, G.; et al. Serial femtosecond crystallography and ultrafast absorption spectroscopy of the photoswitchable fluorescent protein IrisFP. J. Phys. Chem. Lett. 2016, 7, 882–887. [Google Scholar] [CrossRef]
- Krueger, T.D.; Tang, L.; Zhu, L.; Breen, I.L.; Wachter, R.M.; Fang, C. Dual illumination enhances transformation of an engineered green-to-red photoconvertible fluorescent protein. Angew. Chem. Int. Ed. 2020, 59, 1644–1652. [Google Scholar] [CrossRef]
- Kim, H.; Zou, T.; Modi, C.; Dörner, K.; Grunkemeyer, T.J.; Chen, L.; Fromme, R.; Matz, M.V.; Ozkan, S.B.; Wachter, R.M. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure 2015, 23, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Ai, H.-w.; Henderson, J.N.; Remington, S.J.; Campbell, R.E. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem. J. 2006, 400, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Ai, H.-W.; Campbell, R.E. Teal fluorescent proteins: Characterization of a reversibly photoswitchable variant. In Small Animal Whole-Body Optical Imaging Based on Genetically Engineered Probes; SPIE BiOS: San Jose, CA, USA, 2008; p. 68680D. [Google Scholar]
- Stiel, A.C.; Andresen, M.; Bock, H.; Hilbert, M.; Schilde, J.; Schonle, A.; Eggeling, C.; Egner, A.; Hell, S.W.; Jakobs, S. Generation of monomeric reversibly switchable red fluorescent proteins for far-field fluorescence nanoscopy. Biophys. J. 2008, 95, 2989–2997. [Google Scholar] [CrossRef] [Green Version]
- Pletnev, S.; Subach, F.V.; Dauter, Z.; Wlodawer, A.; Verkhusha, V.V. A structural basis for reversible photoswitching of absorbance spectra in red fluorescent protein rsTagRFP. J. Mol. Biol. 2012, 417, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Regis Faro, A.; Carpentier, P.; Jonasson, G.; Pompidor, G.; Arcizet, D.; Demachy, I.; Bourgeois, D. Low-temperature chromophore isomerization reveals the photoswitching mechanism of the fluorescent protein Padron. J. Am. Chem. Soc. 2011, 133, 16362–16365. [Google Scholar] [CrossRef]
- Tiwari, D.K.; Arai, Y.; Yamanaka, M.; Matsuda, T.; Agetsuma, M.; Nakano, M.; Fujita, K.; Nagai, T. A fast- and positively photoswitchable fluorescent protein for ultralow-laser-power RESOLFT nanoscopy. Nat. Methods 2015, 12, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Andresen, M.; Jakobs, S.; Schroeder, J.; Schwarzer, D. Primary light-induced reaction steps of reversibly photoswitchable fluorescent protein Padron0.9 investigated by femtosecond spectroscopy. J. Phys. Chem. B 2015, 119, 5136–5144. [Google Scholar] [CrossRef] [PubMed]
- Konen, T.; Stumpf, D.; Grotjohann, T.; Jansen, I.; Bossi, M.; Weber, M.; Jensen, N.; Hell, S.W.; Jakobs, S. The positive switching fluorescent protein Padron2 enables live-cell reversible saturable optical linear fluorescence transitions (RESOLFT) nanoscopy without sequential illumination steps. ACS Nano 2021, 15, 9509–9521. [Google Scholar] [CrossRef] [PubMed]
- Fron, E.; Van der Auweraer, M.; Hofkens, J.; Dedecker, P. Excited state dynamics of photoswitchable fluorescent protein Padron. J. Phys. Chem. B 2013, 117, 16422–16427. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Tang, L.; Oscar, B.G.; Chen, C. Capturing structural snapshots during photochemical reactions with ultrafast Raman spectroscopy: From materials transformation to biosensor responses. J. Phys. Chem. Lett. 2018, 9, 3253–3263. [Google Scholar] [CrossRef]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein: Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [Green Version]
- Lacombat, F.; Plaza, P.; Plamont, M.-A.; Espagne, A. Photoinduced chromophore hydration in the fluorescent protein Dreiklang is triggered by ultrafast excited-state proton transfer coupled to a low-frequency vibration. J. Phys. Chem. Lett. 2017, 8, 1489–1495. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Tang, L.; Oscar, B.G.; Zhu, L.; Fang, C. Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 2016, 7, 5484–5494. [Google Scholar] [CrossRef] [Green Version]
- Grigorenko, B.L.; Polyakov, I.V.; Krylov, A.I.; Nemukhin, A.V. Computational modeling reveals the mechanism of fluorescent state recovery in the reversibly photoswitchable protein Dreiklang. J. Phys. Chem. B 2019, 123, 8901–8909. [Google Scholar] [CrossRef]
- Sen, T.; Ma, Y.; Polyakov, I.V.; Grigorenko, B.L.; Nemukhin, A.V.; Krylov, A.I. Interplay between locally excited and charge transfer states governs the photoswitching mechanism in the fluorescent protein Dreiklang. J. Phys. Chem. B 2021, 125, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Oscar, B.G.; Liu, W.; Zhao, Y.; Tang, L.; Wang, Y.; Campbell, R.E.; Fang, C. Excited-state structural dynamics of a dual-emission calmodulin-green fluorescent protein sensor for calcium ion imaging. Proc. Natl. Acad. Sci. USA 2014, 111, 10191–10196. [Google Scholar] [CrossRef] [Green Version]
- Pujals, S.; Feiner-Gracia, N.; Delcanale, P.; Voets, I.; Albertazzi, L. Super-resolution microscopy as a powerful tool to study complex synthetic materials. Nat. Rev. Chem. 2019, 3, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Kennis, J.T.M.; Groot, M.-L. Ultrafast spectroscopy of biological photoreceptors. Curr. Opin. Struct. Biol. 2007, 17, 623–630. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Dietze, D.R.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. ChemPhysChem 2016, 17, 1224–1251. [Google Scholar] [CrossRef] [PubMed]
- Kukura, P.; McCamant, D.W.; Yoon, S.; Wandschneider, D.B.; Mathies, R.A. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310, 1006–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, J.; Frontiera, R.R.; Taylor, K.C.; Lagarias, J.C.; Mathies, R.A. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, S.R.; Tang, L.; Wang, Y.; Zhu, L.; Liu, W.; Fang, C. Tuning calcium biosensors with a single-site mutation: Structural dynamics insights from femtosecond Raman spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 7138–7146. [Google Scholar] [CrossRef]
- Konold, P.E.; Arik, E.; Weißenborn, J.; Arents, J.C.; Hellingwerf, K.J.; van Stokkum, I.H.M.; Kennis, J.T.M.; Groot, M.L. Confinement in crystal lattice alters entire photocycle pathway of the photoactive yellow protein. Nat. Commun. 2020, 11, 4248. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, L.; Fang, C. Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales. Int. J. Mol. Sci. 2022, 23, 6459. https://doi.org/10.3390/ijms23126459
Tang L, Fang C. Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales. International Journal of Molecular Sciences. 2022; 23(12):6459. https://doi.org/10.3390/ijms23126459
Chicago/Turabian StyleTang, Longteng, and Chong Fang. 2022. "Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales" International Journal of Molecular Sciences 23, no. 12: 6459. https://doi.org/10.3390/ijms23126459
APA StyleTang, L., & Fang, C. (2022). Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales. International Journal of Molecular Sciences, 23(12), 6459. https://doi.org/10.3390/ijms23126459