
The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis


Abstract
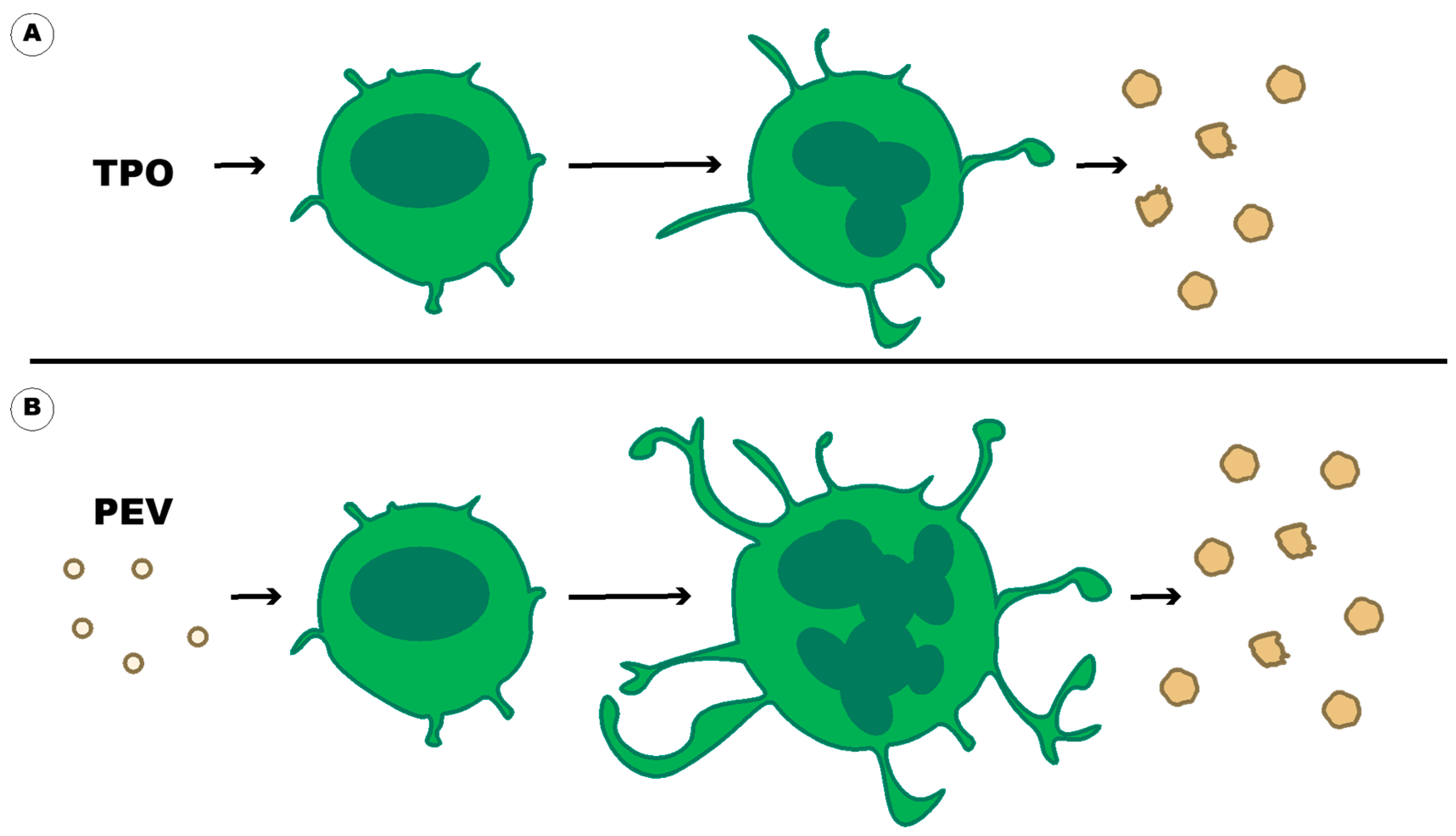
:1. Introduction
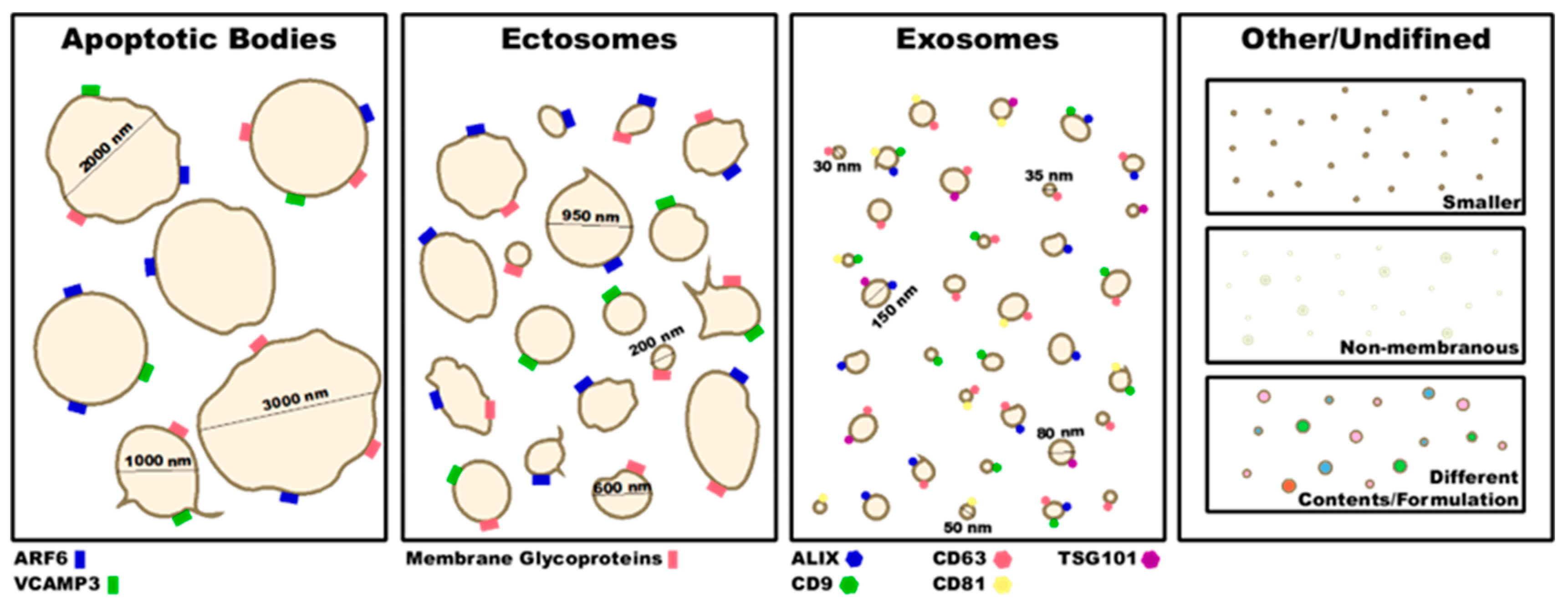
2. Type of EVs and Their Characterization
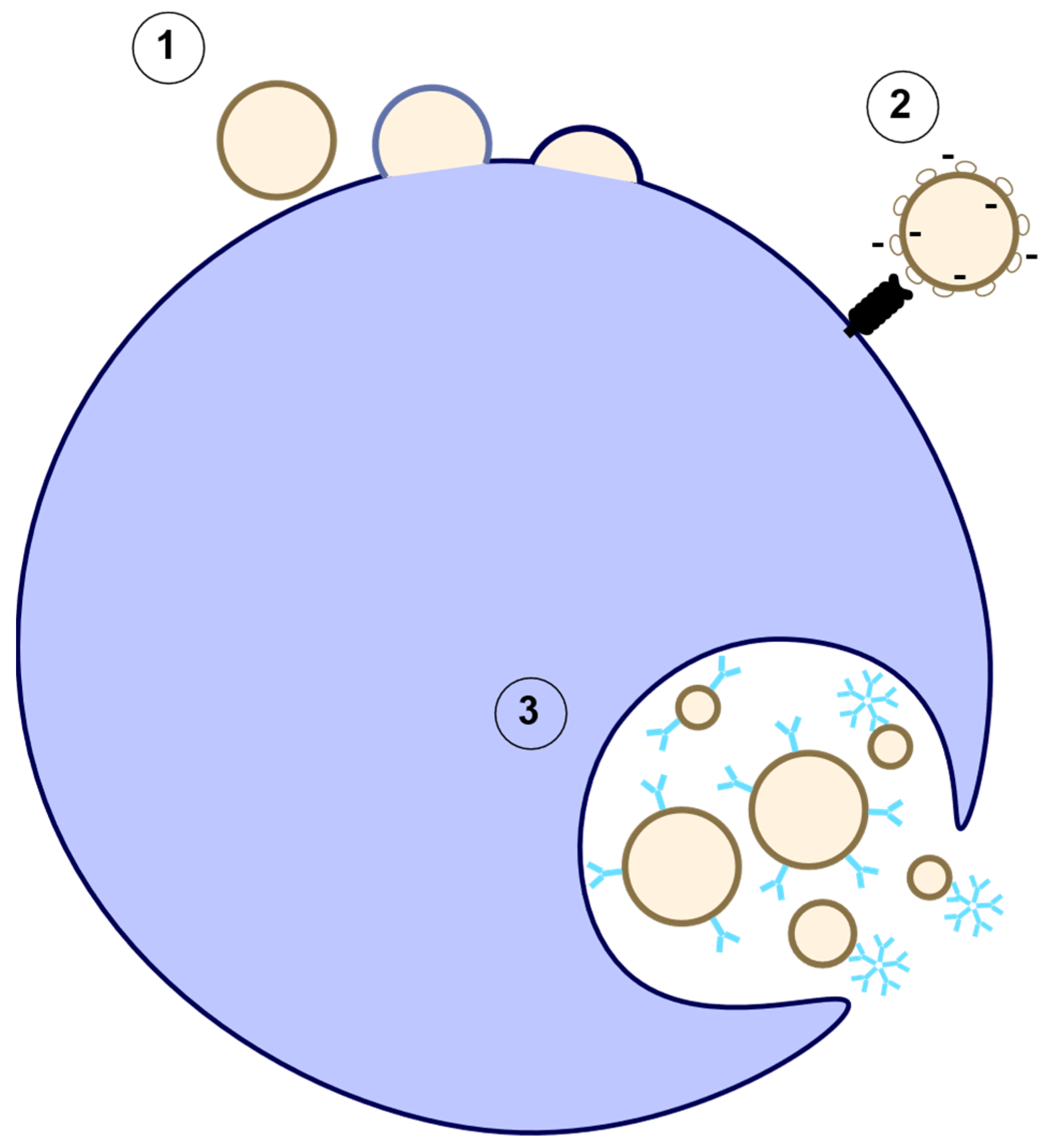
2.1. Exosomes
2.2. Microparticles/Microvesicles
2.3. Apoptotic Bodies
3. EV Characterization
4. EV Clearance
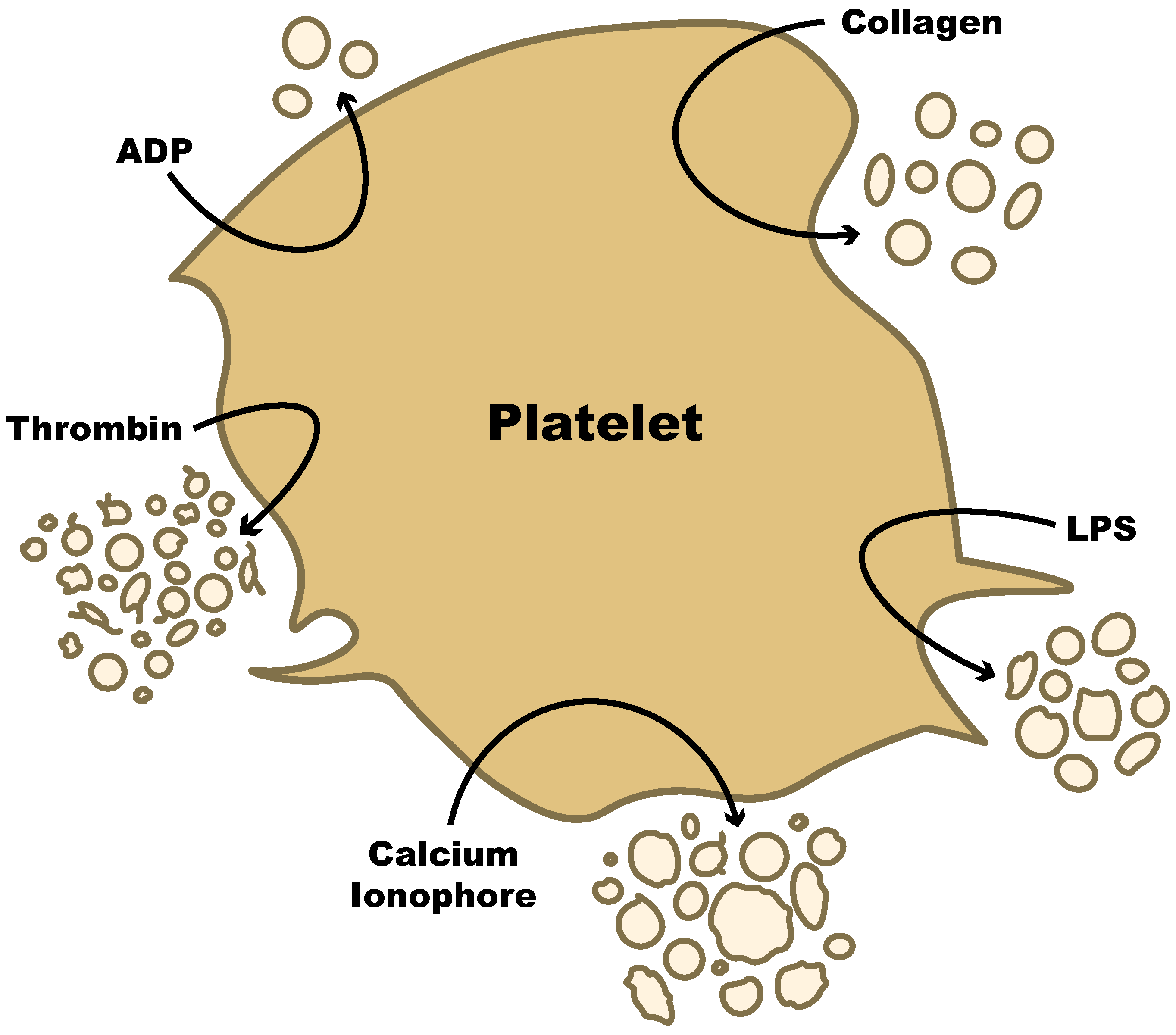
5. Production of Platelet-Derived EVs
5.1. Common Stimuli
5.2. Pathogenic Stimuli
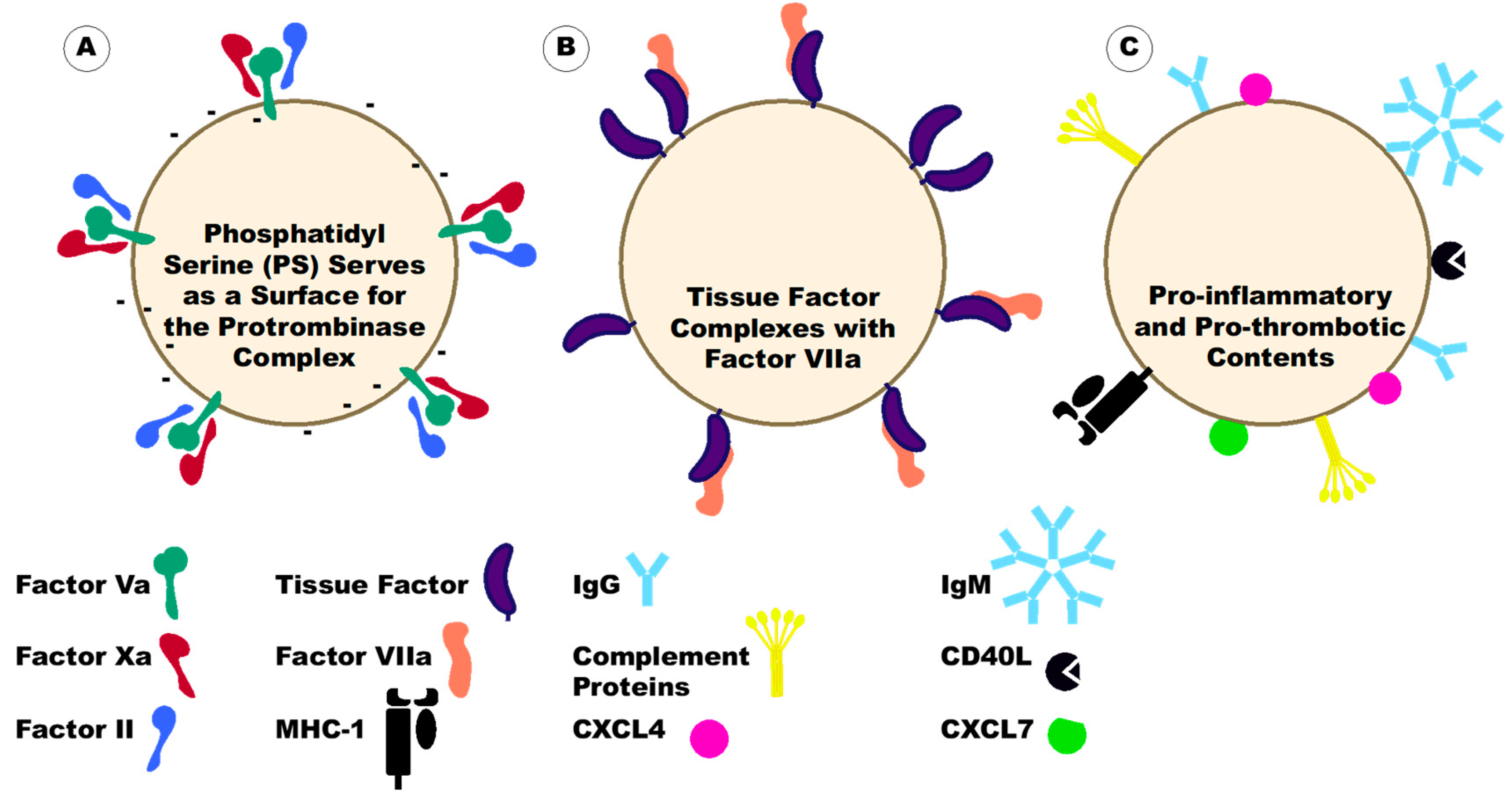
6. Platelet-Derived Extracellular Vesicles under Disease Conditions
7. Platelet-Derived Extracellular Vesicles in Autoimmunity
7.1. Rheumatoid Arthritis (RA)
7.2. Systemic Lupus Erythematosus (Lupus)
7.3. Vasculitis and Systemic Sclerosis
8. Platelet-Derived Extracellular Vesicles in Allergies
9. Platelet-Derived Extracellular Vesicles in Infectious Diseases
9.1. Sepsis
9.2. Human Immunodeficiency Virus (HIV)
9.3. COVID-19
9.4. Dengue
9.5. Malaria
10. Effects of Therapeutics on PEVs
11. A Feedback Loop of Platelet-Derived Extracellular Vesicles Activating Parent Cells
12. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Mairuhu, A.T.A.; Italiano, J.E. Platelet- and megakaryocyte-derived microparticles. Semin. Thromb. Hemos. 2010, 36, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.L.T.; Schmittgen, T.D.; et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaphas, A.; Meyer, J.; Sadoul, K.; Fontana, P.; Morel, P.; Gonelle-Gispert, C.; Buhler, L.H. Platelets and platelet-derived extracellular vesicles in liver physiology and disease. Hepatol. Commun. 2019, 3, 855–866. [Google Scholar] [CrossRef]
- Teixeira, J.; Silva, A.; Almeida, M.I.; Barbosa, M.A.; Santos, S.G. Circulating extracellular vesicles: Their role in tissue repair and regeneration. Trans. Apher. Sci. 2016, 55, 53–61. [Google Scholar] [CrossRef]
- Sabatier, F.; Darmon, P.; Hugel, B.; Combes, V.; Sanmarco, M.; Velut, J.G.; Arnoux, D.; Charpiot, P.; Freyssinet, J.M.; Oliver, C.; et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes 2002, 51, 2840–2845. [Google Scholar] [CrossRef] [Green Version]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef]
- Piguet, P.F.; Kan, C.D.; Vesin, C. Thrombocytopenia in an animal model of malaria is associated with an increased caspase-mediated death of thrombocytes. Apoptosis 2002, 7, 91–98. [Google Scholar] [CrossRef]
- Poveda, E.; Taberbilla, A.; Fitzgerald, W.; Salgado-Barreira, A.; Grandal, M.; Perez, A.; Marino, A.; Alvarez, H.; Valcarce, N.; Gonzalez-Garcia, J.; et al. Massive release of CD9+ microvesicles in human immunodeficiency virus infection, regardless of virologic control. J. Infect. Dis. 2022, 225, 1040–1049. [Google Scholar] [CrossRef]
- Pereira, J.; Alfaro, G.; Goycoolea, M.; Quiroga, T.; Ocqueteau, M.; Massardo, L.; Perez, C.; Saez, C.; Panes, O.; Maus, V.; et al. Circulating platelet-derived microparticles in systemic lupus erythematosus. Association with increased thrombin generation and procoagulant state. Thromb. Haemost. 2006, 95, 94–99. [Google Scholar]
- Thery, C.; Witwer, K.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Simth, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018) a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.; Won, J.; Lim, G.J.; Han, J.; Lee, J.Y.; Cho, K.O.; Bae, Y.K. A novel population of extracellular vesicles smaller than exosomes promotes cell proliferation. Cell Commun. Signal. 2019, 17, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Nevo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021, 12, 4389. [Google Scholar] [CrossRef] [PubMed]
- D’acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Anand, S.; Samule, M.; Mathivanan, S. Exomeres: A new member of extracellular vesicles family. Sub-Cell Biochem. 2021, 97, 89–97. [Google Scholar]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of exosome production and cargo sorting. Int. J. Biol. Sci. 2021, 17, 163–177. [Google Scholar] [CrossRef]
- Oggero, S.; Austin-Williams, S.; Norling, L.V. The contrasting role of extracellular vesicles in vascular inflammation and tissue repair. Front. Pharmacol. 2019, 10, 1479. [Google Scholar] [CrossRef]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling pathways in exosome biogenesis, secretion and fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Debili, N.; Vainchencker, W.; Breton-Gorius, J.; Geuze, H.J.; Sixma, J.J. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood 1998, 91, 2313–2325. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.; Finjheer, R.; Geuze, H.J.; Sixma, G.J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Choezom, D.; Griss, J.C. Neutral sphingomyelinase 2 controls exosome secretion by counteracting V-ATPase-mediated endosome acidification. J. Cell Sci. 2022, 135, jcs259324. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; Van Niel, G.; Kowal, J.; Vignero, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchoir, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Oh, S.; Nacke, M.; Mostov, K.E.; Lipschutz, J.H. Adaptor protein CD2AP and L-type lectin LMAN2 regulate exosome cargo protein trafficking through the Golgi complex. J. Biol. Chem. 2016, 291, 25462–25475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzmann, D.J.; Odorizzi, G.; Emr, S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002, 3, 893–905. [Google Scholar] [CrossRef]
- Cheng, Y.; Schirey, J.S. Targeting soluble proteins to exosomes using a ubiquitin tag. Biotechnol. Bioeng. 2016, 113, 1315–1324. [Google Scholar] [CrossRef]
- Kunadt, M.; Ecjernabb, K.; Stuendl, A.; Gong, J.; Russo, B.; Strauss, K.; Rai, S.; Kugler, S.; Lockhart, L.F.; Schwalbe, M.; et al. Extracellular vesicle sorting of a-Synuclein is regulated by sumoylation. Acta Neuropath. 2015, 129, 695–713. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernandez-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; Baldanta, S.; Enrich, C.; Guerra, S.; Sanchez-Madrid, F. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat. Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Gonzalo, O.; Fernandez-Delgado, I.; Sanchea-Madrid, F. Post-translational add-ons mark the path in exosomal protein sorting. Cell. Mol. Life Sci. 2018, 75, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Harada, Y.; Nakajima, K.; Suzuki, T.; Fukushige, T.; Kondo, K.; Seino, J.; Ohkawa, Y.; Suzuki, T.; Inoue, H.; Kanekura, T.; et al. Glycometabolic regulation of the biogenesis of small extracellular vesicles. Cell Rep. 2020, 33, 108261. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Eng, W.S.; Colquhoun, D.R.; Cinglasan, R.R.; Graham, D.R.; Mahal, L.K. Complex N-linked glycans serve as a determinant for exosome/microvesicle cargo recruitment. J. Biol. Chem. 2014, 289, 32526–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedikter, B.J.; Weseker, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [Green Version]
- Thom, S.R.; Bhopale, V.M.; Yang, M. Neutrophils generate microparticles during exposure to inert gasses due to cytoskeletal oxidative stress. J. Biol. Chem. 2014, 289, 18831–18845. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, L.H.; Cuchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Pare, A.; Rousseau, M.; Naika, G.S.; Levesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Yano, Y.; Kambayashi, J.I.; Shiba, E.; Sakon, M.; Oiki, E.I.J.I.; Fukuda, K.; Kawasaki, T.; Mori, T. The role of protein phosphorylation and cytoskeletal reorganization in microparticle formation from the platelet plasma membrane. Biochem. J. 1994, 299, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Flaumenhaft, R.; Kilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliday, L.S.; De Faria, L.P.; Rody, W.J., Jr. Actin and actin-associated proteins in extracellular vesicles shed by osteoclasts. Int. J. Mol. Sci. 2020, 21, 158. [Google Scholar] [CrossRef] [Green Version]
- Collier, M.E.W.; Maraveyas, A.; Ettelaie, C. Filamin-A is required for the incorporation of tissue factor into cell-derived microvesicles. Thromb. Haemos. 2014, 111, 647–655. [Google Scholar]
- Collier, M.E.W.; Ettelaie, C.; Goult, B.T.; Maraveyas, A.; Goodall, A.H. Investigation of the Filamin A-dependent mechanisms of tissue factor incorporation into microvesicles. Thromb. Haemos. 2017, 117, 2034–2044. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.E.B.; Austin, C.D.; Boyles, J.K.; Steffen, P.K. Role of the membrane skeleton in preventing the shedding of procoagulant-rich microvesicles from the platelet plasma membrane. J. Cell Biol. 1990, 111, 483–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basse, F.; Gaffet, P.; Bienvenue, A. Correlation between inhibition of cytoskeleton proteolysis and anti-vesiculation effect of calpeptin during A23187-induced activation of human platelets: Are vewicles shed by filopod fragmentation? Biochim. Biophys. Acta 1994, 1190, 217–224. [Google Scholar] [CrossRef]
- Cauwenberghs, S.; Feijge, M.; Harper, A.G.S.; Sage, S.O.; Curvers, J.; Heemskerk, J.W.M. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006, 580, 5313–5320. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.E.; Austin, C.D.; Reynolds, C.C.; Steffan, P.K. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J. Biol. Chem. 1991, 266, 13289–13295. [Google Scholar] [CrossRef]
- Wiedmer, T.; Shattil, S.J.; Cunningham, M.; Sims, P.J. Role of calcium and calpain in complement-induced vesiculation of the platelet plasma membrane and in the exposure of the platelet factor Va receptor. Biochemistry 1990, 29, 623–632. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwland, R.; Van Der Pol, E.; Gardiner, C.; Sturk, A. Chapter 22—Platelet-derived microparticles. In Platelets, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 453–467. [Google Scholar]
- Sims, P.J.; Wiedmer, T.; Esmon, C.T.; Weiss, H.J.; Shattil, S.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. J. Biol. Chem. 1989, 264, 17049–17057. [Google Scholar] [CrossRef]
- Mobarrez, F.; Fuzzi, E.; Gunnarsson, I.; Larsson, A.; Eketjall, S.; Pisetsky, D.S.; Svenungsson, E. Microparticles in the blood of patiens with SLE: Size, content of mitochondria and role in circulating immune complexes. J. Autoimmune. 2019, 102, 142–149. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of exosome composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [Green Version]
- Burger, D.; Schock, S.; Thompson, C.S.; Montezano, A.C.; Hakim, A.M.; Touyz, R.M. Microparticles: Biomarkers and beyond. Clin. Sci. 2013, 124, 423–441. [Google Scholar] [CrossRef] [Green Version]
- Arauna, D.; Chiva-Blanch, G.; Padro, T.; Fuentes, E.; Palomo, I.; Badimon, L. Frail older adults show a distinct plasma microvesicle profile suggesting a prothrombotic and proinflammatory phenotype. J. Cell Physiol. 2020, 236, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Terrisse, A.D.; Puech, N.; Allart, S.; Gourdy, P.; Xuereb, J.M.; Payrastre, B.; Sie, P. Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow. J. Thromb. Haemost. 2010, 8, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.L.; Wang, H.; Bang, K.W.A.; Packham, M.A.; Freedman, J. Rapid clearance of procoagulant platelet-derived microparticles from the circulation of rabbits. J. Thromb. Haemost. 2006, 4, 1621–1623. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, A.A.; Nevzorova, T.A.; Mordakhanova, E.R.; Andrianova, I.A.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Intracellular origin and ultrastructure of platelet-derived microparticles. J. Thromb. Haemost. 2017, 15, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Boilard, E.; Duchez, A.C.; Brisson, A. The diversity of platelet microparticles. Curr. Opin. Hematol. 2015, 22, 437–444. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Ullal, A.J.; Gauley, J.; Ning, T.C. Microparticles as mediators and biomarkers of rhumatic disease. Rheumatology 2012, 51, 1737–1746. [Google Scholar] [CrossRef] [Green Version]
- El-Menshawy, N.; Eissa, M.; Farag, R.; Aboalyazed, A. CD235a (Glycophorin-A) is the most predictive value among different circulating cellular microparticles in thrombocytopenic Human Immunodeficiency Virus type 1. J. Clin. Lab Anal. 2016, 30, 235–243. [Google Scholar] [CrossRef]
- Stok, U.; Blokar, E.; Lenassi, M.; Holcar, M.; Frank-Bertoncelj, M.; Erman, A.; Resnik, N.; Sedin-Semrl, S.; Cucnik, S.; Pirkmajer, K.P.; et al. Characterization of plasma-derived small extracellular vesicles indicates ongoing endothelial and platelet activation in patients with thrombotic Antiphospholipid Syndrome. Cells 2020, 9, 1211. [Google Scholar] [CrossRef]
- McCarthy, E.M.; Moreno-Martinez, D.; Wilkinson, F.L.; McHugh, N.J.; Bruce, I.N.; Pauling, J.D.; Alexander, M.Y.; Parker, B. Microparticle subpopulations are potential markers of disease progression and vascular dysfunction across a spectrum of connective tissue disease. Biochim. Biophys. Acta Clin. 2017, 7, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, A.; Veselinovic, M.; Zong, Y.; Jakovljevic, V.; Pruner, I.; Antovic, A. Increased expression of extracellular vesicles is associated with the procoagulant state in patients with established rheumatoid arthritis. Front. Immunol. 2021, 12, 718845. [Google Scholar] [CrossRef]
- Lopez, P.; Rodriguez-Carrio, J.; Martinez-Zapico, A.; Caminal-Montero, L.; Suarez, A. Circulating microparticle subpopulations in Systemic Lupus Erythematosus are affected by disease activity. Int. J. Cardiol. 2017, 236, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Daniel, L.; Fakhouri, F.; Joly, D.; Mouthon, L.; Nusbaum, P.; Grunfeld, J.P.; Schifferli, J.; Guillevin, L.; Lesavre, P.; Halbwachs-Mecarelli, L. Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int. 2006, 69, 1416–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogan, P.A.; Shah, V.; Brachet, C.; Harnden, A.; Many, D.; Klein, N.; Dillon, M.J. Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. 2004, 50, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Taveira-Gomes, T.; Sokhatska, O.; Palmares, C.; Costa, R.; Negrao, R.; Guimaraes, J.T.; Delgado, L.; Soares, R.; Moreira, A. Increased circulating platelet microparticles as a potential biomarker in asthma. Allergy 2013, 68, 1073–1075. [Google Scholar] [CrossRef]
- Punyadee, N.; Mairiang, D.; Thiemmeca, S.; Komoltri, C.; Pan-ngum, W.; Chomanee, N.; Charngkaew, K.; Tangthawornchaikul, N.; Limpitikul, W.; Vasanawathana, S.; et al. Microparticles provide a novel biomarker to predict severe clinical outcomes of Dengue virus infection. J. Virol. 2015, 89, 1587–1607. [Google Scholar] [CrossRef] [Green Version]
- Campello, E.; Radu, C.M.; Simion, C.; Spiezia, L.; Bulato, C.; Gavasso, S.; Tormene, D.; Perin, N.; Turatti, G.; Simioni, P. Longitudinal trend of plasma concentrations of extracellular vesicles in patients hospitalized for COVID-19. Front. Cell Devel. Biol. 2021, 9, 770463. [Google Scholar] [CrossRef]
- Traby, L.; Kussmann, M.; Kussmann, M.; Karer, M.; Sinkovec, H.; Lobmeyr, E.; Hermann, A.; Staudinger, T.; Schellongowski, P.; Rossler, B.; et al. Extracellular vesicles and citrullinated histone H3 in coronavirus disease 2019 patients. Thromb. Haemost. 2022, 122, 113–122. [Google Scholar] [CrossRef]
- Patil, R.; Bajpai, S.; Ghosh, K.; Shetty, S. Microparticles as prognostic biomarkers in dengue virus infection. Acta Trop. 2018, 181, 21–24. [Google Scholar] [CrossRef]
- Mfonkeu, J.B.P.; Gouado, I.; Kuate, H.F.; Zambou, O.; Zollo, P.H.A.; Grau, G.E.R.; Combes, V. Elevated cell-specific microparticles are a biological marker for cerebral dysfunctions in human severe malaria. PLoS ONE 2010, 5, e13415. [Google Scholar]
- Campos, F.M.F.; Franklin, B.S.; Teixeira-Carvalho, A.; Filho, A.L.S.; De Paula, S.C.O.; Fontes, C.J.; Brito, C.F.; Carvalho, L.H. Augmented plasma microparticles during acute Plasmodium vivax infection. Malar. J. 2010, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, C.T.; Ostergaard, O.; Johnsen, C.; Jacobsen, S.; Heegaard, N.H.H. Distinct features of circulating microparticles and their relationship to clinical manifestations in Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Fouda, E.D.E.; Ahmed, M.; Alrayes, M. Circulating endothelial and platelet microparticles for dianosis and monitoring vasculitis. J. Allergy Clin. Immunol. 2014, 133, AB185. [Google Scholar] [CrossRef]
- Leleu, D.; Leviounnois, E.; Laurent, P.; Lazaro, E.; Richez, C.; Duffau, P.; Blanco, P.; Sisirak, V.; Contin-Bordes, C.; Truchetet, M.E. Elevated circulatory levels of microparticles are associated to lung fibrosis and vasculopathy during Systemic Sclerosis. Front. Immunol. 2020, 11, 532177. [Google Scholar] [CrossRef]
- Lehner, G.F.; Harler, U.; Haller, V.M.; Feistritzer, C.; Hasslacher, J.; Dunzedoerfer, S.; Bellmann, R.; Joannidis, M. Characterization of microvesicles in septic shock using high-sensitivity flow cytometry. Shock 2016, 46, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijmans, J.G.; Stockelman, K.; Garcia, V.; Levy, M.V.; Brewster, L.M.; Bammert, T.D.; Greiner, J.J.; Stauffer, B.L.; Connick, E.; DeSouza, C.A. Circulating microparticles are elevated in treated HIV-1 infection and are deleterious to endothelial cell function. J. Am. Heart Assoc. 2019, 8, e011134. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; El-Badawy, O.; Ali, W.A.; Mahran, Z.G.; Mahran, E.E.M.O.; Rayan, A. Circulating microparticles and activated platelets as novel prognostic biomarkers in COVID-19; relation to cancer. PLoS ONE 2021, 16, e0246806. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, O.; Nielsen, C.T.; Tanassi, J.T.; Iversen, L.V.; Jacobsen, S.; Heegaard, N.H.H. Distinct proteome pathology of circulating microparticles in Systemic Lupus Erythematosus. Clin. Proteom. 2017, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Boscolo, A.; Campello, E.; Bertini, D.; Spiezia, L.; Lucchetta, V.; Piasentini, E.; Radu, C.M.; Manesso, L.; Ori, C.; Simioni, P. Levels of circulating microparticles in septic shock and sepsis-related complications: A case-control study. Minerva Anetesiol. 2019, 85, 625–634. [Google Scholar] [CrossRef]
- Manzano, E.M.; Fernandez-Bello, I.; Sanz, R.J.; Marhuenda, A.R.; Lopez-Longo, F.J.; Acuna, P.; Roman, M.T.A.; Yuste, V.J.; Butta, N.V. Insights into the procoagulant profile of patients with Systemic Lupus Erythematosus without antiphospholipid antibodies. J. Clin. Med. 2020, 9, 3297. [Google Scholar] [CrossRef]
- Rondina, M.T.; Tatsumi, K.; Bastarache, J.A.; Mackman, N. Microvesicle tissue factor activity and interleukin-8 levels are associated with mortality in patients with Influenza A/H1N1 infection. Crit. Care Med. 2016, 44, e574–e578. [Google Scholar] [CrossRef]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; et al. Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv. 2021, 5, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Ovstebo, R.; Hellum, M.; Aass, H.C.D.; Troseid, A.M.; Brandtzaeg, P.; Mollnes, T.E.; Henriksson, C.E. Microparticle-associated tissue factor activity is reduced by inhibition of the complement protein 5 in Neisseria meningitidis-exposed whole blood. Innate Immun. 2014, 20, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, A.; Takahashi, Y.; Chang, H.Y.; Wu, Y.W.; Yamamoto, A.; Ishihama, Y.; Takakura, Y. Blood concentrations of small extracellular vesicles are determined by a balance between abundant secretion and rapid clearance. J. Extracell. Vesicles 2019, 9, 1696517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcoux, G.; Laricge, A.; Hasse, S.; Bellio, M.; Mbarik, M.; Tamagne, M.; Allaeys, I.; Zufferey, A.; Levesque, T.; Robetz, J.; et al. Platelet EVs contain an active proteasome involved in protein processing for antigen presentation via MHC-1 molecules. Blood 2021, 138, 2607–2620. [Google Scholar] [CrossRef]
- Rank, A.; Nieuwland, R.; Crispin, A.; Grutzner, S.; Iberer, M.; Toth, B.; Pihusch, R. Clearance of Platelet Microparticles In Vivo. Platelets 2011, 22, 111–116. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Huber, L.C.; Hueber, A.J.; Reich, C.F., III; Gay, S.; Distler, O.; Pisetsky, D.S. The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis 2005, 10, 731–741. [Google Scholar] [CrossRef]
- Dasgupta, S.K.; Abdel-Monem, H.; Niravath, P.; Le, A.; Bellera, R.V.; Langlois, K.; Nagata, S.; Rumbaut, R.E.; Thiagarajan, P. Lactadherin and clearance of platelet-derived microvesicles. Blood 2008, 113, 1332–1339. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Cai, W.; Zhao, Z.; Hilton, T.; Wang, M.; Yeon, J.; Liu, W.; Zhang, F.; Shi, F.D.; Wu, X.; et al. Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of severe TBI mice. Blood 2018, 131, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Litvack, M.L.; Post, M.; Palaniyar, N. IgM promotes the clearance of small particles and apoptic microparticles by macrophages. PLoS ONE 2011, 6, e17223. [Google Scholar]
- Bilyy, R.O.; Shkandina, T.; Tomin, A.; Munoz, L.E.; Franz, S.; Antonyuk, V.; Kit, Y.Y.; Zirngibl, M.; Furnrohr, B.G.; Janko, C.; et al. Macrophages discriminate glycosylation patterns of apoptotic cell-derived microparticles. J. Biol. Chem. 2012, 287, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Yao, Q.P.; Huang, K.; Chen, X.H.; Han, Y.; Jiang, Z.L.; Gao, L.Z.; Qi, Y.X. Platelet-Derived miR-142-3p Induces Apoptosis of Endothelial Cells in Hypertension. Cell Mol. Biol. 2017, 63, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, P.; Culibrk, L.; Culibrk, B.; Conway, E.M.; Goodrich, R.P.; Devine, D.V. Releasates of riboflavin/UV-treated platelets: Microvesicles suppress cytokine-mediated endothelial cell migration/proliferation. Transfusion 2021, 61, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nomura, S.; Miyake, T.; Kagawa, H.; Kitada, C.; Taniguchi, H.; Komiyama, Y.; Fujimura, Y.; Ikeda, Y.; Fukuhara, S. High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles. Blood 1996, 88, 3456–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, T.W.; Hellums, J.D.; Thiagarajan, P. Thrombin receptor activating peptide (SFLLRN) potentiates shear-induced platelet microvesiculation. J. Lab. Clin. Med. 2000, 135, 66–72. [Google Scholar] [CrossRef]
- Jiang, J.; Woulge, D.D.; Papoutsakis, E.T. Shear enhances thrombopoiesis and formation of microparticles that induce megakaryocytic differentiation of stem cells. Blood 2014, 124, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Rui, S.; Yuan, Y.; Du, C.; Song, P.; Chen, Y.; Wang, H.; Fan, Y.; Armstrong, D.G.; Deng, W.; Li, L. Comparison and investigation of exosomes derived from platelet-rich plasma activated by different agonists. Cell Transplant. 2021, 30, 09636897211017833. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Sahler, J.; Kim, N.; Spinelli, S.L.; Maggirwar, S.B.; Garraud, O.; Cognasse, F.; Blumberg, N.; Phipps, R.P. Breaking the mold: Transcription factors in the anucleate platelet and platelet-derived microparticles. Front. Immuno. 2015, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.M.; Nasirikenari, M.; Manhardt, C.T.; Ashline, D.J.; Hanneman, A.J.; Reinhold, V.N.; Lau, J.T.Y. Platelets support extracelluler sialyation by supplying the sugar donor substrate. J. Biol. Chem. 2014, 289, 8742–8748. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, R.S.; Ferreira, P.M.; Mitchell, J.L.; Pula, G.; Gibbins, J.M. Platelet-derived extracellular vesicles express NADPH osidase-1 (NOX-1), generate superoxide and modulate platelet function. Free Radic. Biol. Med. 2021, 165, 395–400. [Google Scholar] [CrossRef]
- Yin, W.G.B.; Peerschke, E.I.B. Expression of complement components and inhibitors on platelet microparticles. Platelets 2008, 19, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.S.; Huang, T.; Hsieh, S.L. Extracellular vesicles from CLEC-2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.T.; McIntyre, T. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinglammatory IL-1B-Rich Microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Gambim, M.; Carmo, A.O.; Marti, L.; Verissimo-Filho, S.; Lopes, L.R.; Janiszewski, M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: Experimental evidence for a novel mechanism of septic vascular dysfunction. Crit. Care 2007, 11, R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bei, J.J.; Liu, C.; Peng, S.; Liu, C.H.; Zhao, W.B.; Qu, X.L.; Chen, Q.; Zhou, Z.; Yu, Z.P.; Peter, K.; et al. Staphylococcal SSL5-induced platelet microparticles provoke proinflammatory responses via the CD40/TRAF6,NFkB signalling pathway in monocytes. Thromb. Haemost. 2016, 115, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Gama, W.M.; Oliveira, L.B.; Chaves, Y.O.; Ribeiro, F.; Almeida, T.V.R.; Baptista, B.J.A.; Santana, M.F.; Ferreira, L.C.; Lacerda, M.V.G.; Nogueira, P.A. Increased levels of reactive oxygen species in platelets and platelet-derived microparticles and the risk of respiratory failure in HIV/AIDS patients. Mem. Inst. Oswaldo 2020, 115, e200082. [Google Scholar] [CrossRef]
- Zhang, W.L.J.; Liang, J.; Qi, X.; Tian, J.; Liu, J. Coagulation in lymphatic system. Front. Cardiovasc. Med. 2021, 8, 762648. [Google Scholar] [CrossRef]
- Cantadori, L.O.; Gaiolla, R.; Niero-Melo, L.; Oliveira, C.C. Bone marrow aspirate clot: A useful technique in diagnosis of follow-up of hematological disorders. Case Rep. Hematol. 2019, 2019, 7590948. [Google Scholar] [CrossRef]
- Dalens, B.; Bezou, M.; Coulet, M.; Haberer, J.P.; Vanneuville, G. Spinal fluid clotting activity: A new method of evaluating neonatal brain damage. Pediatr. Res. 1982, 16, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Graeber, J.E.; Stuart, M.J. Spinal-fluid procoagulant activity: A sensitive indicator of central-nervous-system damage. Lancet 1978, 312, 285–288. [Google Scholar] [CrossRef]
- Niewiarowski, S.; Jausmanowa-Petrusewicz, I.; Wegrzynowicz, Z. Blood clotting factors in cerebrospinal fluid. J. Clin. Pathol. 1962, 15, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Stirling, P.; Faroug, R.; Freemont, T. Anticoagulating synovial fluid samples in septic arthritis. Rheumatology 2014, 53, 2315–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermansen, P.; Freemont, T. Synovial fluid analysis in the diagnosis of joint disease. Diagnos. Histopathol. 2017, 23, 211–220. [Google Scholar] [CrossRef]
- Carmassi, F.; Negri, F.; Morale, M.; Puccetti, R.; Song, K.Y.; Chung, S.I. Assessment of coagulation and fibrinolysis in synovial fluid of rheumatoid arthritis patients. Fibrinolysis 1994, 8, 162–171. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.; Cervellin, G. Hemostatic properties of the lymph: Relationships with occlusion and thrombosis. Semin. Thromb. Hemos. 2012, 38, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Opie, E.L. Thrombosis and occlusion of lymphatics. J. Med. Res. 1913, 29, 131–146. [Google Scholar] [PubMed]
- Johansson, K.; Chong, H.; Ciornei, C.D.; Brorson, H.; Mortimer, P.S. Axillary web syndrome: Evidence for lymphatic origin with thrombosis. Lymphat. Res. Biol. 2019, 18, 329–332. [Google Scholar] [CrossRef]
- Hara, H.; Mihara, M.; Ohtomo, R.; Tanaka, S. Lymphatic vessel thrombosis in a patient with secondary lymphedema. Plast. Reconstr. 2019, 7, e2268. [Google Scholar] [CrossRef]
- Bao, H.; Chen, Y.; Huang, K.; Zhuang, F.; Bao, M.; Han, Y.; Chen, X.H.; Shi, Q.; Yao, Q.P.; Qi, Y.X. Platelet-derived microparticles promote endothelial cell proliferation in hypertension via miR-142-3p. FASEB J. 2018, 32, 3912–3923. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovid, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef]
- Chimen, M.; Evryviadou, A.; Box, C.L.; Harrison, M.J.; Haxeldine, J.; Dib, L.H.; Kuravi, S.J.; Payne, H.; Price, J.M.J.; Kavanagh, D.; et al. Appropriation of GPIba from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica 2020, 105, 1248–1261. [Google Scholar] [CrossRef]
- Zubairova, L.D.; Nabiullina, R.; Nagaswami, C.; Zuev, Y.F.; Mustafin, I.G.; Litvinov, R.I.; Weisel, J.W. Circulating microparticles alter formation, structure, and properties of fibrin clots. Sci. Rep. 2015, 5, 17611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet extracellular vesicles. Arterioscler. Thromb. Vasc. Biol. 2021, 47, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.R.; Alexander, W.; Hassoune, A.; Chen, Q.; Brzoska, T.; Alvikas, J.; Liu, Y.; Haldeman, S.; Plautz, W.; Loughran, P.; et al. Platelet-derived extracellular vesicles released after trauma promote hemostasis and contribute to DVT in mice. J. Thromb. Haemost. 2019, 17, 1733–1745. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Kireev, D.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [PubMed]
- Somajo, S.; Koshiar, K.L.; Norstrom, E.; Dahlback, B. Protein S and factor V in regulation of coagulation on platelet microparticles by activated protein C. Thromb. Res. 2014, 134, 144–152. [Google Scholar] [CrossRef]
- Schwertz, H.; Tolley, N.; Foulks, J.M.; Denis, M.M.; Risenmay, B.W.; Buerke, M.; Tilley, R.E.; Rondina, M.T.; Harris, E.M.; Kraiss, L.W.; et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenecity of human platelets. J. Exp. Med. 2006, 203, 2433–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondina, M.T.; Schwerz, H.; Harris, E.S.; Kraemer, B.F.; Campbell, R.A.; Mackman, N.; Grissom, C.K.; Weyrich, A.S.; Zimmerman, G.A. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J. Thromb. Haemost. 2011, 9, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matus, V.; Valenzuela, J.; Hidalgo, P.; Pozo, L.M.; Panes, O.; Wozniak, A.; Mezzano, D.; Pereira, J.; Saez, C.G. Human platelet interaction with E. coli O111 promotes tissue-factor-dependent procoagulant activity, involving Toll like receptor 4. PLoS ONE 2017, 12, e0185431. [Google Scholar] [CrossRef]
- Bouchard, B.A.; Gissel, M.; Whelihan, M.F.; Mann, K.G.; Butenas, S. Platelets do not express the oxidized or reduced forms of tissue factor. Biochim. Biophys. Acta 2014, 1840, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- Osterud, B.; Olsen, J.O. Human platelets do not express tissue factor. Thromb. Res. 2013, 132, 112–115. [Google Scholar] [CrossRef]
- Del Conde, I.; Shrimpton, C.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Villar-Vesga, J.; Grajales, C.; Burbano, C.; Vanegas-Garcia, A.; Munoz-Vahos, C.H.; Vasquez, G.; Rojas, M.; Castano, D. Platelet-derived microparticles generated in vitro resemble circulating vesicles of patients with rheumatoid arthritis and activate monocytes. Cell. Immunol. 2019, 336, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthrisis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Luo, L.; Morgelin, M.; Thorlacius, H. Rac1 regulates sepsis-induced formation of platelet-derived microparticles and thrombin generation. Biochem. Biophys. 2017, 487, 887–891. [Google Scholar] [CrossRef]
- Wang, W.; Deng, Z.; Liu, G.; Yang, J.; Zhou, W.; Zhang, C.; Shen, W.; Zhang, Y. Platelet-derived extracellular vesicles promote the migration and invasion of rheumatoid arthritis fibroblast-like synoviocytes via CXCR2 signaling. Exp. Ther. Med. 2021, 22, 1120. [Google Scholar] [CrossRef]
- Panigrahi, S.; Ghosh, S.K.; Ferrari, B.; Wyrick, J.M.; Podrez, E.A.; Weinberg, A.; Sieg, S.F. Human B-defensin-3 is associated with platelet-derived extracellular vesicles and is a potential contributor to endothelial dysfunction. Front. Mol. Bio. 2022, 9, 824954. [Google Scholar] [CrossRef]
- Szilagyi, B.; Fejes, Z.; Rusznyak, A.; Fenyvesi, F.; Pocsi, M.; Halmi, S.; Griger, Z.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B., Jr. Platelet microparticles enriched in miR-223 reduce ICAM-1-dependent vascular inflammation in septic conditions. Front. Physiol. 2021, 12, 658524. [Google Scholar] [CrossRef]
- Janiszewski, M.; Do Carmo, A.O.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R.M. Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: A novel vascular redox pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef]
- Baker, J.V.; Brummel-Ziedins, K.; Neuhaus, J.; Duprez, D.; Cummins, N.; Dalmau, D.; De Hovitz, J.; Lehmann, C.; Sullivan, A.; Woolley, I.; et al. HIV replication alters the composition of extrinsic pathway coagulation factors and increases thrombin generation. J. Am. Heart Assoc. 2013, 2, e000264. [Google Scholar]
- Rozmyslowicz, T.; Majka, M.; Kijowdki, J.; Murphy, S.L.; Conover, D.O.; Poncz, M.; Ratajczak, J.; Gaulton, G.N.; Ratajczak, M.Z. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 2003, 17, 33–42. [Google Scholar] [CrossRef]
- Rausch, L.; Lutz, K.; Schifferer, M.; Winheim, E.; Gruber, R.; Oesterhaus, E.F.; Rinke, L.; Hellmuth, J.C.; Scherer, C.; Muenchhoff, M.; et al. Binding of phosphatidylserine-positive microparticles by PBMCs classifies disease severity in COVID-19 patients. J. Extracell. Vesicles 2021, 10, e12173. [Google Scholar] [CrossRef] [PubMed]
- Garnier, Y.; Claude, L.; Hermand, P.; Sachou, E.; Claes, A.; Despoan, K.; Chahim, B.; Roger, P.M.; Martino, F.; Colin, Y.; et al. Plasma microparticles of intubated COVID-19 patients cause endothelial cell death, neutrophil adhesion and netosis, in a phosphatidylserine-dependent manner. Br. J. Haematol. 2021, 196, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Lopes, J.; Freitas, C.; Valls-de-Souza, R.; Oliveira, M.F.; Bozza, M.T.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faille, D.; Combes, V.; Mitchell, A.J.; Fontaine, A.; Juhan-Vague, I.; Alessi, M.C.; Chimini, G.; Fusai, T.; Grau, G.E. Platelet microparticles: A new player in malaria parasite cytoadherence to human brain endothelium. FASEB J. 2009, 23, 3449–3458. [Google Scholar] [CrossRef]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, A.; Cassidy, H.; Ainle, F.N.; Caprodossi, A.; Lanuti, P.; et al. Platelet-derived microparticles from obese individuals: Characterizatino of number, size, proteomics and crosstalk with cancer and endothelial cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Miao, D.; Ma, T.T.; Chen, M.; Zhao, M.H. Platelets release proinflammatory microparticles in anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatology 2019, 58, 1432–1442. [Google Scholar] [CrossRef]
- Andoh, A.; Tsujikawa, T.; Hata, K.; Araki, Y.; Kitoh, K.; Sasaki, M.; Yoshida, T.; Fujiyama, Y. Elevated circulating platelet-derived microparticles in patients with active inflammatory bowel disease. Am. J. Gastroenterol. 2005, 100, 2042–2048. [Google Scholar] [CrossRef]
- Sheremata, W.A.; Jy, W.; Horstman, L.L.; Ahn, Y.S.; Alexander, J.S.; Minagar, A. Evidence of platelet activation in multiple sclerosis. J. Neuroinflamm. 2008, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Tomczynska, M.; Slata, I.; Bijak, M.; Saluk-Bijak, J. The potential contribution and role of a blood platelets in autoimmune thyroid diseases. J. Cell Mol. Med. 2018, 22, 6386–6390. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Ostergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H.H. Increased IgG on cell-derived plasma microparticles in Systemic Lupus Erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar]
- Liao, T.L.; Hsieh, S.; Chen, Y.M.; Chen, H.H.; Liu, H.J.; Lee, H.C.; Chen, D.Y. Rituximab may cause increased Hepatitis C virus uiremia in Rheumatoid Arthritis patients through declining exosomal MicroRNA-155. Arthritis Rheum. 2018, 70, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Knijff-Dutmer, E.A.; Koerts, J.; Nieuwland, R.; Kalsbeek-Batenburg, E.M.; Van de Laar, M.A.F.J. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Ni, J.; Witherel, C.E.; Yang, M.; Burdick, J.A.; Wen, C.; Wong, S.H.D. Harnessing tissue-derived extracellular vesicles for osteoarthritis theranostics. Theranostics 2022, 12, 207–231. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Capra, J.; Rilla, K.; Lehenkari, P.; Oikari, S.; Kaariainen, T.; Joukainen, A.; Kroger, H.; Paakkonen, T.; Matilainen, J.; et al. Characterization of hyaluronan-coated extracellular vesicles in synovial fluid of patient with osteoarthritis and rheumatoid arthritis. BMC Musculoskelet. Disord. 2021, 22, 247. [Google Scholar] [CrossRef] [PubMed]
- Tessandier, N.; Melki, I.; Cloutier, N.; Allaeys, I.; Miszta, A.; Tan, S.; Milasan, A.; Michel, S.; Benmoussa, A.; Levesque, T.; et al. Platelets disseminate extracellular vesicles in lymph in Rheumatoid Arthritis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 929–942. [Google Scholar] [CrossRef]
- Lood, C.; Tyden, H.; Gullstrand, B.; Nielsen, C.T.; Heegaard, N.H.H.; Linge, P.; Jonsen, A.; Hesselstrand, R.; Kahn, R.; Bengtsson, A.A. Decreased platelet size is associated with platelet activation and anti-phospholipid syndrome in Systemic Lupus Erythematosus. Rheumatology 2017, 56, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Mobarrez, F.; Vikerfors, A.; Gustafsson, J.T.; Gunnarsson, I.; Zickert, A.; Larsson, A.; Pisetsky, D.S.; Wallen, H.; Svenungsson, E. Microparticles in the blood of patients with systemic lupus erythematosus (SLE): Phenotypic characterization and clinical associations. Sci. Rep. 2016, 6, 36025. [Google Scholar] [CrossRef]
- Burbano, C.; Villar-Vesga, J.; Orejuela, J.; Munoz, C.; Vanegas, A.; Vasquez, G.; Rojas, M.; Castano, D. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 322. [Google Scholar] [CrossRef]
- Ostergaard, O.; Nielsen, C.T.; Iversen, L.V.; Tanassi, J.T.; Knudsen, S.; Jacobsen, S.; Heegaard, N.H.H. Unique protein signiture of circulating microparticles in Systemic Lupus Erythematosus. Arthritis Rheum. 2013, 65, 2680–2690. [Google Scholar]
- Fortin, P.R.; Cloutier, N.; Bissonnette, V.; Aghdassi, E.; Eder, L.; Simonyan, D.; Laflamme, N.; Boilard, E. Distinct subtypes of microparticle-containing immune complexes are associated with disease activity, damage, and carotid intima-media thickness in Systemic Lupus Erythematosus. J. Rheumatol. 2016, 43, 2019–2025. [Google Scholar] [CrossRef]
- Miao, D.; Li, D.Y.; Chen, M.; Zhao, M.H. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res. Ther. 2017, 19, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiducci, S.; Distler, J.; Jungel, A.; Huscher, D.; Huber, L.C.; Michel, B.A.; Gay, R.E.; Pisetsky, D.S.; Gay, S.; Matucci-Cerinic, M.; et al. The relationship between plasma microparticles and disease manifestations in patients with systemic sclerosis. Arthritis Rheum. 2008, 58, 2845–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, S.; Norihito, I.; Ozaki, Y.; Kagawa, H.; Fukuhara, S. Significance of microparticles in progressive systemic sclerosis with interstitial pneumonia. Platelets 2008, 19, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Kayaleh, B.; Denton, C.P.; Mason, J.C.; Matucci-Cerinic, M. ANCA in systemic sclerosis, when vasculitis overlaps with vasculopathy: A devastating combination of pathologies. Rheumatology 2021, 60, 5509–5516. [Google Scholar] [CrossRef]
- Manfredi, A.A.; Ramirez, G.A.; Godino, C.; Capobianco, A.; Monno, A.; Franchini, S.; Tombetti, E.; Corradetti, S.; Distler, J.H.W.; Bianchi, M.E.; et al. Platelet phagocytosis via P-selectin glycoprotein ligand 1 and accumulation of microparticles in Systemic Sclerosis. Arthritis Rheum. 2022, 74, 318–328. [Google Scholar] [CrossRef]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Valle, P.D.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Trans. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Capobianco, S.; Takabayashi, T.; Schleimer, R.P. Chronic Rhinosinusitis and the coagulation system. Allergy Asthma Immunol. 2015, 7, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Shimizu, T.; Morser, J.; Kobayashi, T.; Yamaguchi, A.; Qin, L.; Toda, M.; D-Alessandro-Gabazza, C.; Maruyama, T.; Takagi, T.; et al. Role of the coagulation system in allergic inflammation in the upper airways. Clin. Immunol. 2008, 129, 365–371. [Google Scholar] [CrossRef]
- Guilarte, M.; Sala-Cunill, A.; Luengo, O.; Labrador-Horrillo, M.; Cardona, V. The mast cell, contact, and coagulation system connection in anaphylaxis. Front. Immunol. 2017, 8, 846. [Google Scholar] [CrossRef] [Green Version]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Seasonal changes in platelet activity in pollen-induced seasonal allergic rhinitis and asthma. J. Asthma 2008, 45, 485–487. [Google Scholar] [CrossRef]
- Tamagawa-Mineoka, R.; Katoh, N.; Ueda, E.; Masuda, K.; Kishimoto, S. Platelet-derived microparticles and soluble P-selectin as platelet activation markers in patients with atopic dermatitis. Clin. Immunol. 2009, 131, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sprague, D.L.; Elzey, B.D.; Crist, S.A.; Waldschmidt, T.J.; Jensen, R.J.; Ratliff, T.L. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 2008, 111, 5028–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Zhu, T.; Liu, J.; Guo, Z.; Cao, X. Platelets promote allergic asthma through the expression of CD154. Cell. Mol. Immunol. 2015, 12, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, M.T.; Zurlo, C.; Bon, M.; Bertomoro, A.; Bendo, R.; Bertozzi, I.; Radu, C.M.; Campello, E.; Simioni, P.; Fabris, F. Platelet-derived microparticles bearing PF4 and Anti-GAGS Immunoglobulins in patients with sepsis. Diagnostics 2020, 10, 627. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The role of extracellular vesicles in viral infection and transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Pleet, M.L.; DeMarino, C.D.; Stonier, S.W.; Dye, J.M.; Jacobson, S.; Aman, M.J.; Kashanchi, F. Extracellular vesicles and Ebola virus: A new mechanism of immune evasion. Viruses 2019, 11, 410. [Google Scholar] [CrossRef] [Green Version]
- Arakelyan, A.; Fitzgerald, W.; Zicari, S.; Vanpouille, C.; Margolis, L. Extracellular vesicles carry HIV Env and facilitate HIV infection of human lymphoid tissue. Sci. Rep. 2017, 7, 1695. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Schierer, S.; Blume, K.; Dindorf, J.; Wittki, S.; Xiang, W.; Ostalecki, C.; Koliha, N.; Wild, S.; Schuler, G.; et al. HIV-Nef and ADAM17-containing plasma extracellular vesicles induce and correlate with immune pathogenesis in chronic HIV infection. EBioMedicine 2016, 6, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Coakley, G.; Maizels, R.; Buck, A.H. Exosomes and other extracellular vesicles: The new communicators in parasite infections. Trends Parasitol. 2015, 31, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Sisquella, X.; Ofir-Birin, Y.; Pimentel, M.A.; Cheng, L.; Karam, P.A.; Sampaio, N.G.; Penington, J.S.; Connolly, D.; Giladi, T.; Scicluna, B.J.; et al. Malaria parasite DNA-harbouring vesicles activate cytosolic immune sensors. Nat. Commun. 2017, 8, 1985. [Google Scholar] [CrossRef]
- Shimizu, M.; Konihi, A.; Nomura, S. Examination of biomarker expressions in sepsis-related DIC patients. Int. J. Gen. Med. 2018, 11, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puskarich, M.A.; Cornelius, D.C.; Bandyopadhyay, S.; McCalmon, M.; Tramel, R.; Dale, W.D.; Jones, A.E. Phosphatidylserine expressiong platelet microparticle levels at hospital presentation are decreased in spesis non-survivors and correlate with thrombocytopenia. Thromb. Res. 2008, 168, 138–144. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Luo, L.; Norstrom, E.; Braun, O.O.; Morgelin, M.; Thorlacius, H. Platelet-derived microparticles regugulates thrombin generation via phosphatidylserine in abdominal sepsis. J. Cell. Physiol. 2018, 233, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Tokes-Fuzesi, M.; Woth, G.; Ernyey, B.; Vermes, I.; Muhl, D.; Bogar, L.; Kovacs, G.L. Microparticles and acute renal dysfunction in septic patients. J. Crit. Care 2013, 28, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Woth, G.; Tokes-Fuzesi, M.; Magyarlaki, T.; Kovacs, G.L.; Vermes, I.; Muhl, D. Activated platelet-derived microparticle numbers are elevated in patients with severe fungal (Candida albicans) sepsis. Ann. Clin. Biochem. 2012, 49, 554–560. [Google Scholar] [CrossRef]
- Machado, G.B.S.; De Assis, M.C.; Leao, R.; Saliba, A.M.; Silva, M.C.A.; Suassuna, J.H.; De Oliveira, A.V.; Plotkowski, M.C. Exon-induced vascular hyperpermeability and platelet activation in the course of experimental Pseudomonas aeruginosa pneumosepsis. Shock 2010, 33, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Pare, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Levesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcyRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Funderburg, N.T.; Lederman, M.M. Coagulation and morbidity in treated HIV infection. Thromb. Res. 2014, 133, S21–S24. [Google Scholar] [CrossRef] [Green Version]
- Fink, A.; Knudsen, A.D.; Thudium, R.F.; Von Stemann, J.H.; Afzul, S.; Lundgren, J.; Kirkegaard-Klitbo, D.M.; Ostrowski, S.R.; Nordestgaard, B.G.; Nielsen, S.D. Identification of two different coagulation phenotypes in people living with HIV with undetectable viral replication. Sci. Rep. 2021, 11, 4383. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Nardi, M.A.; Li, Z. HIV-1 Tat induced-platelet activation and release of CD154 contribute to HIV-1 associated autoimmune thrombocytopenia. J. Thromb. Haemost. 2011, 9, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Falasca, K.; Lanuti, P.; Ucciferri, C.; Pieragostino, D.; Cufaro, M.C.; Bologna, G.; Federici, L.; Miscia, S.; Pontolillo, M.; Auricchio, A.; et al. Circulating extracellular vesicles as new inflammation marker in HIV infection. AIDS 2021, 35, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.; Portier, I.; Rowley, J.; Stubben, C.; Petrey, A.; Tolley, N.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in COVID-19 Patients. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Catello, L.; et al. Circulating platelet-derived extracellular vesicles are a hallmark of SARS-CoV-2 infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Maugeri, N.; De Lorenzo, R.; Clementi, N.; Diotti, R.A.; Criscuolo, E.; Godino, C.; Tresoldi, C.; Study Group BAfCB; Bonini, C.; Clementi, M.; et al. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J. Thromb. Haemost. 2022, 20, 434–448. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; El Haj, R.B.; et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Abdelmaksoud, M.F.; Abdelmaksoud, S.S.; Abdelsamee, H.F.; Ezzelregal, H.G.; Alfeky, M.A. Platelets derived microparticles in COVID-19: Correlation to inflammatory and coagulation state. J. Appl. Hematol. 2021, 12, 195–202. [Google Scholar]
- Kaur, S.; Singh, A.; Kaur, J.; Verma, N.; Pandey, A.K.; Das, S.; Bhattacharyya, S.; Guchhait, P. Upregulation of cytokine signalling in platelets increases risk of thrombophilia in severe COVID-19 patients. Blood Cell Mol. Dis. 2022, 94, 102653. [Google Scholar] [CrossRef]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating exosomes are strongly involved in SARS-CoV-2 Infection. Front. Mol. Bio. 2021, 8, 632290. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Bury, L.; Camilloni, B.; Castronari, R.; Piselli, E.; Malvestiti, M.; Borghi, M.; KuchiBotla, H.; Falcinelli, E.; Petito, E.; Amato, F.; et al. Search for SARS-CoV-2 RNA in platelets from COVID-19 patients. Platelets 2021, 32, 284–287. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, J.; Fang, Y.; Lu, S.; Wu, J.; Zheng, X.; Deng, F. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J. Hematol. Oncol. 2021, 14, 72. [Google Scholar] [PubMed]
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets. J. Thromb. Haemost. 2021, 19, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal. Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Wills, B.A.; Oragui, E.; Stephens, A.C.; Daramola, O.A.; Dung, N.M.; Loan, H.T.; Chau, N.V.; Chambers, M.; Stepniewska, K.; Farrar, J.J.; et al. Coagulation abnormalities in dengue hemorrhagic fever: Serial investigations in 167 Vietnamese children with Dengue shock syndrome. Clin. Infect. Dis. 2002, 32, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Sailo, L.; Pradhan, D.; Nongthombam, R.; Bhattacharyya, P. Disseminated intravascular coagulation in malaria: A case report. Niger. Med. J. 2014, 55, 171–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iso-o, N.; Komatsuya, K.; Tokumasu, F.; Isoo, N.; Ishigaki, T.; Yasui, H.; Yotsuyanagi, H.; Hara, M.; Kita, K. Malaria parasites hijcak host receptors from exosomes to capture lipoproteins. Front. Cell Devel. Biol. 2021, 9, 749153. [Google Scholar] [CrossRef]
- Toda, H.; Diaz-Varela, M.; Segui-Barber, J.; Roobsoong, W.; Baro, B.; Garcia-Silva, S.; Galiano, A.; Gualdron-Lopez, M.; Almeida, A.C.G.; Brito, M.A.M.; et al. Plasma-derived extracellular vesicles from Plasmodium vivax patients signal spleen fibroblasts via NF-kB facilitating parasite cytoadherence. Nat. Commun. 2020, 11, 2761. [Google Scholar] [CrossRef]
- Otero, R.; Elias, T.; Montes-Werboys, A.; Dawson, G.; Lampa, R.; Jara, L.; Scully, M. Effects of long-term anticoagulant therapy on levels of circulating microparticles in patients with deep venous thrombosis. Blood Coagul. Fibrinolysis 2011, 22, 628–629. [Google Scholar] [CrossRef]
- Chyrchel, B.; Drozdz, A.; Dlugosz, D.; Stepien, E.L.; Surdacki, A. Platelet reactivity and circulating platelet-derived microvesicles are differentially affected by P2Y12 receptor antagonists. Int. J. Mol. Sci. 2019, 16, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Goubran, H.; Seghatchian, J.; Sabry, W.; Ragab, G.; Burnouf, T. Platelet and extracellular vesicles in COVID-19 infection and its vaccines. Transfus. Apher. Sci. 2022, 61, 103459. [Google Scholar] [CrossRef]
- Bouayard, A. HLA and PF4 antibody production after adenoviral vector SARS-CoV-2 vaccination. Curr. Res. Transl. Med. 2021, 69, 103312. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, T.A.; Mordakhanova, E.R.; Daminova, A.G.; Pnomareva, A.A.; Andrianova, L.A.; Le Minh, G.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Platelet factor 4-containing immune complexes induce platelet activation followed by calpain-dependent platelet death. Cell Death Discov. 2019, 5, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, M.; Zou, X.; Fang, F.; Wang, S.; Xu, L.; Zeng, Q.; Fan, Z.; Chen, L.; Yue, W.; Xie, X.; et al. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat. Commun. 2020, 11, 4964. [Google Scholar] [CrossRef] [PubMed]
- French, S.L.; Butov, K.; Allaeys, I.; Canas, J.; Morad, G.; Davenport, P.; Laroche, A.; Trubina, N.M.; Italiano, J.E., Jr.; Moses, M.A.; et al. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020, 4, 3011–3023. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Raslova, H. Megakaryocyte polyploidizatino: Role in platelet production. Platelets 2020, 31, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Shin, J.; Wang, Y.; Min, S.H.; Poncz, M.; Choi, J.K.; Discher, D.E.; Carpenter, C.L.; Lian, L.; Zhao, L.; et al. RhoA is essential for maintaining normal megakaryocyte ploidy and platelet generation. PLoS ONE 2013, 8, e69315. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Koh, G.Y. Platelets take the lead in lymphatic separation. Circ. Res. 2010, 106, 1184–1186. [Google Scholar] [CrossRef] [Green Version]
- Antich-Rossello, M.; Forteza-Genestra, M.A.; Monjo, M.; Ramis, J.M. Platelet-derived extracellular vesicles for regenerative medicine. Int. J. Mol. Sci. 2021, 22, 8580. [Google Scholar] [CrossRef]
- Lopez, E.; Srivastava, A.; Burchfield, J.; Wang, Y.W.; Cardenas, J.C.; Togarrati, P.P.; Miyazawa, B.; Gonzalez, E.; Holcomb, J.B.; Pati, S.; et al. Platelet-derived extracellular vesicles promote hemostasis and prevent the development of hemorrhagic shock. Sci. Rep. 2019, 9, 17676. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell of Origin | Protein Marker | References |
|---|---|---|
| Platelet | CD41, CD42b, CD61, CD62P | [2,3,5,9,37,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71] |
| Megakaryocyte | CD41, CD42b, filamin A | [2,5,6,37] |
| Red Blood Cell | Glycophorin A (CD235) | [5,7,58,66,69,70,71] |
| Endothelial Cell | CD62E, CD31, CD105, CD144, CD146 | [5,53,57,59,60,61,62,64,67,68,69,70,71,72,73,74,75,76,77] |
| Neutrophil | CD16, CD66b | [3,6,63] |
| Monocyte | CD14, CD11b | [3,5,6,9,53,58,61,70,71,76,78] |
| Leukocyte | CD45 | [5,6,59,61,67,68,71,72,76,79] |
| T-Cell | CD3, CD4, CD8 | [3,5,9,62] |
| B-Cell | CD19, CD20, CD79a | [3,5] |
| NK Cell | CD56 | [67,80,81,82] |
| Prothrombotic | CD142 (tissue factor) | [10,39,54,83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eustes, A.S.; Dayal, S. The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis. Int. J. Mol. Sci. 2022, 23, 7837. https://doi.org/10.3390/ijms23147837
Eustes AS, Dayal S. The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis. International Journal of Molecular Sciences. 2022; 23(14):7837. https://doi.org/10.3390/ijms23147837
Chicago/Turabian StyleEustes, Alicia S., and Sanjana Dayal. 2022. "The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis" International Journal of Molecular Sciences 23, no. 14: 7837. https://doi.org/10.3390/ijms23147837
APA StyleEustes, A. S., & Dayal, S. (2022). The Role of Platelet-Derived Extracellular Vesicles in Immune-Mediated Thrombosis. International Journal of Molecular Sciences, 23(14), 7837. https://doi.org/10.3390/ijms23147837