
A Novel Morphological Parameter Predicting Fibrotic Evolution in Myeloproliferative Neoplasms: New Evidence and Molecular Insights

 ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
1.1. MPNs’ Molecular Landscape, In Vivo and In Vitro Models and Possible Novel Therapeutic Strategies
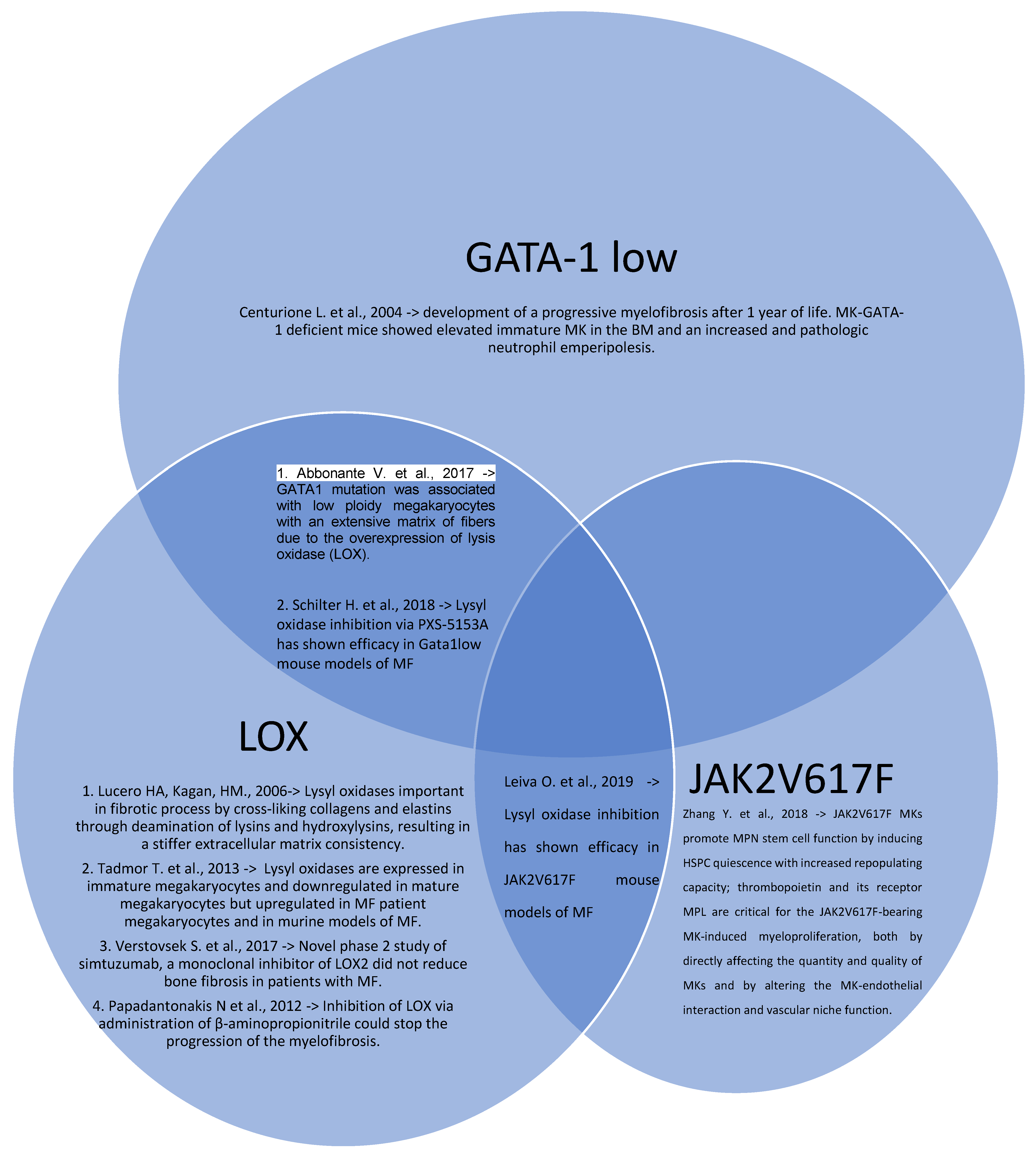
1.2. GATA-1 Low Models
1.3. Lysyl Oxidases Models
1.4. Profibrotic Agent Models

1.5. In Vitro Models

{kind=link}
| List of In Vitro Models | Outcomes |
|---|---|
| Larocca L.M.; Heller P.G.; Podda G. et al. [26] “Megakaryocytic emperipolesis and platelet function abnormalities in five patients with gray platelet syndrome.” | Cultures of CD34+ HSCs from patients with fibrotic MPNs proved that MKs overly expanded, were immature and escaped death signal. |
| Martyré, M.C. et al. [27] “Elevated levels of basic fibroblast growth factor in megakaryocytes and platelets from patients with idiopathic myelofibrosis.” | Expression of mutant JAK2 in megakaryocytes was sufficient to induce fibrosis and erythropoiesis, the latter due to increased levels of IL6 and other cytokines such as IL-1β. |
| Teofili L. et al. [28] “Endothelial progenitor cell dysfunction in myelodysplastic syndromes: possible contribution of a defective vascular niche to myelodysplasia.” | Mimicking of the MDS and MPN vascular via ECFC, which express less CD34, CD41, AML1 and GPIb, thus impeding the normal megakaryocytic differentiation and/or maturation. |
| Villeval J.L.; Cohen-Solal K.; Tulliez M. et al. [29] “High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice.” | In vitro cultures with basic fibroblast growth factor (bFGF) present in megakaryocytes showed that it was not exported into the medium, consistent with the fact that bFGF is devoid of a secretion peptide signal. |
| Psaila, Bethan et al. [30] “Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets.” | Single-cell RNA sequencing megakaryocyte-biased hematopoiesis in myelofibrosis showed that aberrant megakaryopoiesis in IMF is due to both aberrant differentiation of HSPCs as well as proliferation of mature megakaryocytes. |
| Coxon C.H.; Geer M.J.; Senis Y.A. [31] “ITIM receptors: more than just inhibitors of platelet activation.” | MK from IMF patients aberrant metabolic and inflammatory signatures. |
| Senis Y.A.; Tomlinson M.G.; García A.; Dumon S.; Heath V.L.; Herbert J.; Cobbold S.P.; Spalton J.C.; Ayman S.; Antrobus R. [32] “A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein.” | MK from IMF patients harbor some aberrant surface markers expression, in particular G6B, an immunoreceptor exclusively found on mature MKs. |
| Becker, Isabelle C. et al. [33] “G6b-B regulates an essential step in megakaryocyte maturation.” | MPIG6B-mutated were smaller in size, displayed a less-developed demarcation membrane system and reduced expression of receptors. RNA sequencing proved an overall reduction of megakaryocyte-specific transcripts, as well as decreased protein levels of GATA-1, and impaired thrombopoietin signaling. Increased neutrophil emperipolesis into mutant MKs in situ by transmission electron microscopy (TEM) and in cryosections was also observed. |
1.6. Therapeutic Agents
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S. Diagnostic criteria and prognosis in polycythemia vera and essential thrombocythemia. Semin. Hematol. 1999, 36, 9–13. [Google Scholar] [PubMed]
- Georgii, A.; Buesche, G.; Kreft, A. The histopathology of chronic myeloproliferative diseases. Baillieres Clin. Haematol. 1998, 11, 721–749. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Schino, M.; Fiorentino, V.; Rossi, E.; Betti, S.; Di Cecca, M.; Ranucci, V.; Chiusolo, P.; Martini, M.; De Stefano, V.; Larocca, L.M. Bone marrow megakaryocytic activation predicts fibrotic evolution of Philadelphia-negative myeloproliferative neoplasms. Hematologica 2021, 106, 3162. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and karyotype- enhanced international prognostic scoring system for primary myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Abbonante, V.; Di Buduo, C.A.; Gruppi, C.; Malara, A.; Gianelli, U.; Celesti, G.; Anselmo, A.; Laghi, L.; Vercellino, M.; Visai, L.; et al. Thrombopoietin/TGF-b1 Loop Regulates Megakaryocyte extracellular Matrix Component Synthesis. Stem Cells 2016, 34, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Woods, B.; Chen, W.; Chiu, S.; Marinaccio, C.; Fu, C.; Gu, L.; Bulic, M.; Yang, Q.; Zouak, A.; Jia, S.; et al. Activation of JAK/STAT signaling in megakaryocytes sustains myeloproliferation in vivo. Clin. Cancer Res. 2019, 25, 5901–5912. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, S.; Kramer, F.; Mullally, A.; Lane, S.W. Murine Models of Myelofibrosis. Cancers 2020, 12, 2381. [Google Scholar] [CrossRef]
- Centurione, L.; Di Baldassarre, A.; Zingariello, M.; Bosco, D.; Gatta, V.; Rana, R.A.; Langella, V.; Di Virgilio, A.; Vannucchi, A.M.; Migliaccio, A.R. Increased and pathologic emperipolesis of neutrophils within megakaryocytes associated with marrow fibrosis in GATA-1 (low) mice. Blood 2004, 104, 3573–3580. [Google Scholar] [CrossRef] [Green Version]
- Abbonante, V.; Chitalia, V.; Rosti, V.; Leiva, O.; Matsuura, S.; Balduini, A.; Ravid, K. Upregulation of lysyl oxidase and adhesion to collagen of human megakaryocytes and platelets in primary myelofibrosis. Blood 2017, 130, 829–831. [Google Scholar] [CrossRef] [Green Version]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The lysyl oxidase like 2/3 enzymatic inhibitor, PXS-5153A, reduces crosslinks and ameliorates fibrosis. J. Cell Mol. Med. 2018, 23, 1759–1770. [Google Scholar] [CrossRef] [Green Version]
- Leiva, O.; Ng, S.K.; Matsuura, S.; Chitalia, V.; Lucero, H.; Findlay, A.; Turner, C.; Jarolimek, W.; Ravid, K. Novel lysyl oxidase inhibitors attenuate hallmarks of primary myelofibrosis in mice. Int. J. Hematol. 2019, 110, 699–708. [Google Scholar] [CrossRef]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Dong, H.; Maltzman, J.D.; Sharma, S.; Silverman, J.; Oh, S.T.; Gotlib, J. A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Brit. J. Haematol. 2017, 176, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Papadantonakis, N.; Matsuura, S.; Ravid, K. Megakaryocyte pathology and bone marrow fibrosis: The lysyl oxidase connection. Blood 2012, 120, 1774–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential trombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malara, A.; Abbonante, V.; Zingariello, M.; Migliaccio, A.; Balduini, A. Megakaryocyte Contribution to Bone Marrow Fibrosis: Many Arrows in the Quiver. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lin, C.H.S.; Kaushansky, K.; Zhan, H. JAK2V617F megakaryocytes promote hematopoietic stem/progenitor cell expansion in mice through thrombopoietin/MPL signaling. Stem Cells 2018, 36, 1676–1684. [Google Scholar] [CrossRef] [Green Version]
- Larocca, L.M.; Heller, P.G.; Podda, G.; Pujol-Moix, N.; Glembotsky, A.C.; Pecci, A.; Alberelli, M.A.; Balduini, C.L.; Landolfi, R.; Cattaneo, M.; et al. Megakaryocytic emperipolesis and platelet function abnormalities in five patients with gray platelet syndrome. Platelets 2015, 26, 751–757. [Google Scholar] [CrossRef]
- Martyré, M.-C.; Le Bousse-Kerdiles, M.-C.; Romquin, N.; Chevillard, S.; Praloran, V.; Demory, J.-L.; Dupriez, B. Elevated levels of basic fibroblast growth factor in megakaryocytes and platelets from patients with idiopathic myelofibrosis. Br. J. Haematol. 1997, 97, 441–448. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Nuzzolo, E.R.; Capodimonti, S.; Iachininoto, M.G.; Cocomazzi, A.; Fabiani, E.; Voso, M.T.; Larocca, L.M. Endothelial progenitor cell dysfunction in myelodysplastic syndromes: Possible contribution of a defective vascular niche to myelodysplasia. Neoplasia 2015, 17, 401–409. [Google Scholar] [CrossRef]
- Villeval, J.L.; Cohen-Solal, K.; Tulliez, M.; Giraudier, S.; Guichard, J.; Burstein, S.A.; Cramer, E.M.; Vainchenker, W.; Wendling, F. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood 1997, 90, 4369–4383. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Wang, G.; Rodriguez-Meira, A.; Li, R.; Heuston, E.F.; Murphy, L.; Yee, D.; Hitchcock, I.S.; Sousos, N.; O’Sullivan, J.; et al. Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets. Mol. Cell 2020, 78, 477–492.e8. [Google Scholar] [CrossRef]
- Coxon, C.H.; Geer, M.J.; Senis, Y.A. ITIM receptors: More than just inhibitors of platelet activation. Blood 2017, 129, 3407–3418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senis, Y.A.; Tomlinson, M.G.; García, A.; Dumon, S.; Heath, V.L.; Herbert, J.; Cobbold, S.P.; Spalton, J.C.; Ayman, S.; Antrobus, R.; et al. A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol. Cell. Proteom. 2007, 6, 548–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, I.C.; Nagy, Z.; Manukjan, G.; Haffner-Luntzer, M.; Englert, M.; Heib, T.; Vögtle, T.; Gross, C.; Bharti, R. G6b-B regulates an essential step in megakaryocyte maturation. Blood Adv. Bloodadv. 2022, 8, 3155–3161. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. JAK inhibitors for myeloproliferative neoplasms: Clarifying facts from myths. Blood 2012, 119, 2721–2730. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Hoffman, R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood 2013, 121, 4832–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J.; Verstovsek, S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat. Rev. Drug Discov. 2011, 10, 127–140. [Google Scholar] [CrossRef]
- Mascarenhas, J. Selective Janus associated kinase 1 inhibition as a therapeutic target in myelofibrosis. Leuk. Lymphoma 2015, 56, 2493–2497. [Google Scholar] [CrossRef]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.; et al. Primary Analysis of a Phase II Open-Label Trial of INCB039110, a Selective JAK1 Inhibitor, in Patients with Myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Goldenson, B.; Silver, S.J.; Schenone, M.; Dancik, V.; Huang, Z.; Wang, L.Z.; Lewis, T.A.; An, W.F.; Li, X.; et al. Identification of regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell 2012, 150, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Q.J.; Yang, Q.; Goldenson, B.; Malinge, S.; Lasho, T.; Schneider, R.K.; Breyfogle, L.J.; Schultz, R.; Gilles, L.; Koppikar, P.; et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat. Med. 2015, 21, 1473–1480. [Google Scholar] [PubMed]
- Sumi, K.; Tago, K.; Kasahara, T.; Funakoshi-Tago, M. Aurora kinase A critically contributes to the resistance to anti-cancer drug cisplatin in JAK2 V617F mutant-induced transformed cells. FEBS Lett. 2011, 585, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Gangat, N.; Marinaccio, C.; Swords, R.; Watts, J.M.; Gurbuxani, S.; Rademaker, A.; Fought, A.J.; Frankfurt, O.; Altman, J.K.; Wen, Q.J.; et al. Aurora Kinase A Inhibition Provides Clinical Benefit, Normalizes Megakaryocytes, and Reduces Bone Marrow Fibrosis in Patients with Myelofibrosis: A Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4898–4906. [Google Scholar] [CrossRef] [Green Version]
- Lima, K.; Carlos, J.A.E.G.; Alves-Paiva, R.; Vicari, H.P.; de Souza Santos, F.P.; Hamerschlak, N.; Costa-Lotufo, L.V.; Traina, F.; Machado-Neto, J.A. Reversine exhibits antineoplastic activity in JAK2V617F-positive myeloproliferative neoplasms. Sci. Rep. 2019, 9, 9895. [Google Scholar] [CrossRef]
- Ceglia, I.; Dueck, A.C.; Masiello, F.; Martelli, F.; He, W.; Federici, G.; Petricoin, E.F., III; Zeuner, A.; Iancu-Rubin, C.; Weinberg, R.; et al. Preclinical rationale for TGF-β inhibition as a therapeutic target for the treatment of myelofibrosis. Exp. Hematol. 2016, 44, 1138–1155.e1134. [Google Scholar] [CrossRef] [Green Version]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [Green Version]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y., Jr.; Levine, R.L.; et al. LSD1 inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere 2018, 2, e54. [Google Scholar] [CrossRef]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 29–43.e7. [Google Scholar] [CrossRef] [Green Version]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.N.; Oh, S.T. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Palandri, F.; Sabattini, E.; Maffioli, M. Treating early-stage myelofibrosis. Ann. Hematol. 2019, 98, 241–253. [Google Scholar] [CrossRef] [PubMed]
| Prognostic Model and Risk Factors (Weight) | Risk Groups and Median Survival | |
|---|---|---|
| IPSS | ||
| Hemoglobin < 10 g/dL (1 point) | Low risk: 0 point (135 months) | |
| Leukocytes > 25 × 109/L (1 point) | Intermediate risk-1:1 point (95 months) | |
| Age > 65 years (1 point) | Intermediate risk-2:2 points (48 months) | |
| Circulating blast ≥ 1% (1 point) | High risk: ≥3 points (27 months) | |
| Constitutional symptoms (1 point) | ||
| DIPSS. Same variables as IPSS, apart from: | ||
| Hemoglobin < 10 g/dL (2 points) | ||
| Low risk: 0 point (not reached) | ||
| Intermediate risk-1:1–2 points (14.2 yrs) | ||
| Intermediate risk-2:3–4 points (4 yrs) | ||
| High risk: 5–6 points (1.5 yrs) | ||
| DIPSS+. Same variables of DIPSS, apart from: | ||
| Unfavorable karyotype (1 point) | Low risk: 0 point (185 months) | |
| Red cell transfusion need (1 point) | Intermediate risk-1:1 point (78 months) | |
| Hemoglobin < 10 g/dL (1 point) | Intermediate risk-2:2–3 points (35 months) | |
| Platelet < 100 × 109/L (1 point) | High risk: 4–6 points (16 months) | |
| Prognostic model and risk factors (weight) | Risk groups and median survival | |
| MIPSS70. Same variables as DIPSS+, apart from: | ||
| Genetic variables | Clinical variables | |
| One high molecular risk (HMR) mutation (1 point) | Marrow fibrosis grade ≥ 2 (1 point) | Low risk: 0–1 point (not reached) |
| ≥2 HMR mutations (2 points) | Leukocytes > 25 × 109/L (2 points) | Intermediate risk: 2–4 (6.3 yr) |
| Type 1/like CALR absent (1 point) | Platelet < 100 × 109/L (2 points) | High risk: ≥5 (3.1 yr) |
| Circulating blast ≥ 2% (1 point) | ||
| MIPSS70+ version 2.0 | ||
| Genetic variables | Clinical variables | |
| VHR karyotype (4 points) | Severe anemia (2 points) | Very low risk: 0 point (not reached) |
| Unfavorable karyotype (3 points) | Moderate anemia (1 point) | Low risk: 1–2 (16.4 yr) |
| ≥2 HMR mutations (3 points) | Circulating blasts ≥ 2% (1 point) | Intermediate-1 risk: 3–4 (7.7 yr) |
| One HMR mutation (2 points) | Constitutional symptoms (2 points) | High risk: 5–8 (4.1 yr) |
| Type 1/like CALR absent (2 points) | Very high risk: ≥9 (1.8 yr) | |
| GIPSS. Based on a genetic-only risk factors model. | ||
| VHR karyotype (2 points) | Low risk: 0 point (26.4 yr) | |
| Unfavorable karyotype (1 point) | Intermediate-1 risk: 1 point (8 yr) | |
| Type 1/like CALR absent (1 point) | Intermediate-2 risk: 2 points (4.2 yr) | |
| ASXL1 mutation (1 point) | High risk: ≥3 points (2 yr) | |
| SRSF2 mutation (1 point) | ||
| U2AF1Q157 mutation (1 point) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorentino, V.; Tralongo, P.; Martini, M.; Betti, S.; Rossi, E.; Pierconti, F.; De Stefano, V.; Larocca, L.M. A Novel Morphological Parameter Predicting Fibrotic Evolution in Myeloproliferative Neoplasms: New Evidence and Molecular Insights. Int. J. Mol. Sci. 2022, 23, 7872. https://doi.org/10.3390/ijms23147872
Fiorentino V, Tralongo P, Martini M, Betti S, Rossi E, Pierconti F, De Stefano V, Larocca LM. A Novel Morphological Parameter Predicting Fibrotic Evolution in Myeloproliferative Neoplasms: New Evidence and Molecular Insights. International Journal of Molecular Sciences. 2022; 23(14):7872. https://doi.org/10.3390/ijms23147872
Chicago/Turabian StyleFiorentino, Vincenzo, Pietro Tralongo, Maurizio Martini, Silvia Betti, Elena Rossi, Francesco Pierconti, Valerio De Stefano, and Luigi Maria Larocca. 2022. "A Novel Morphological Parameter Predicting Fibrotic Evolution in Myeloproliferative Neoplasms: New Evidence and Molecular Insights" International Journal of Molecular Sciences 23, no. 14: 7872. https://doi.org/10.3390/ijms23147872
APA StyleFiorentino, V., Tralongo, P., Martini, M., Betti, S., Rossi, E., Pierconti, F., De Stefano, V., & Larocca, L. M. (2022). A Novel Morphological Parameter Predicting Fibrotic Evolution in Myeloproliferative Neoplasms: New Evidence and Molecular Insights. International Journal of Molecular Sciences, 23(14), 7872. https://doi.org/10.3390/ijms23147872