
Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes

 and
and
Abstract
:1. Introduction
2. Brain Energy Metabolism
2.1. Glucose Metabolic Routes
2.2. The Important Role of Astrocytes in Brain Energy Metabolism
2.3. Astrocyte–Neuron Lactate Shuttle
3. Brain Energy Metabolism in Cerebral Ischemia
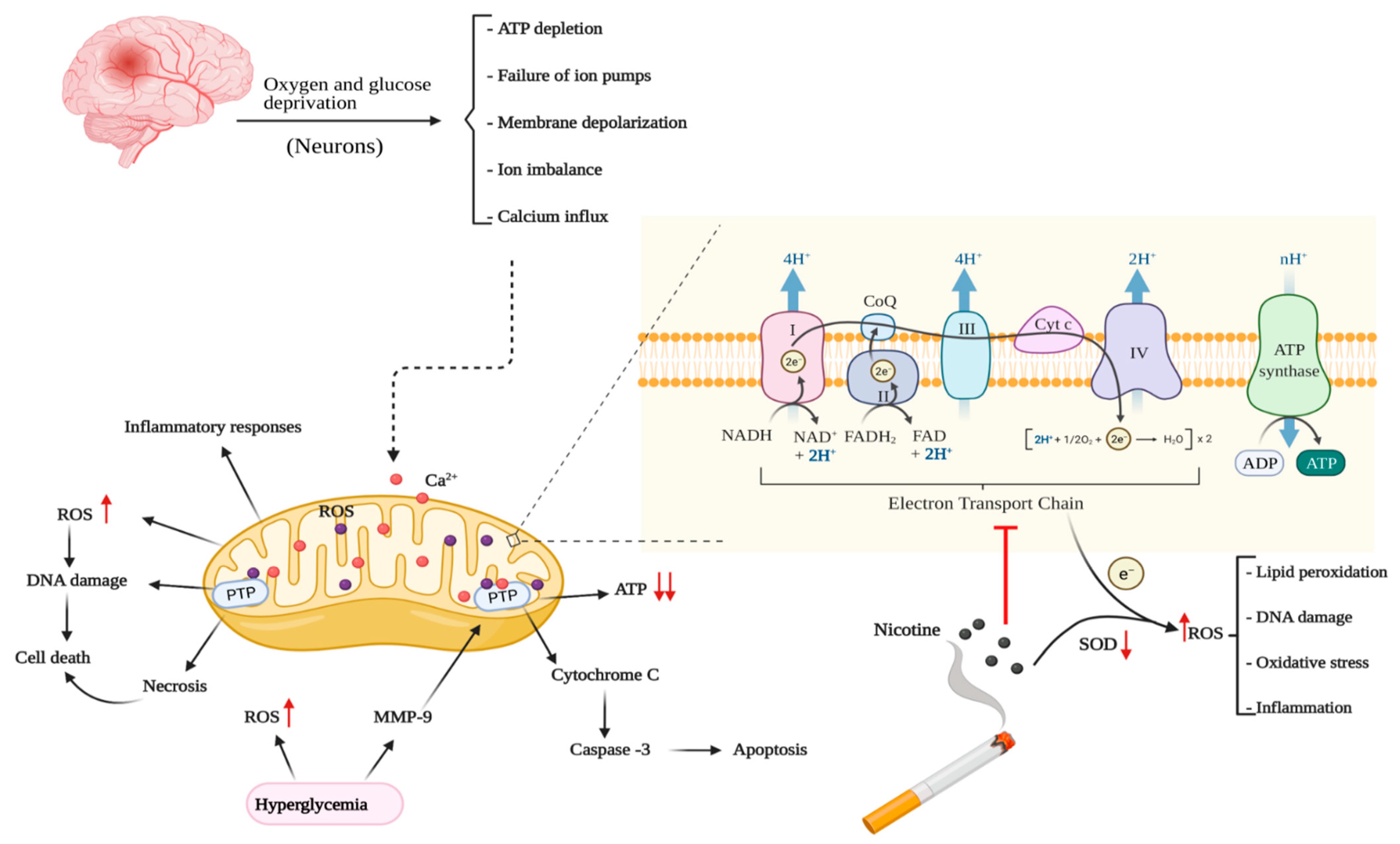
3.1. Role of Mitochondria in Ischemic Brain Energy Metabolism
3.2. Sources of Oxidative Stress in Cerebral Ischemia
3.3. Metabolic Flexibility of Microglia: Potential Role in Ischemic Stroke
4. Smoking and Diabetes as Comorbid Conditions for Ischemic Stroke
5. Effects of Nicotine and Smoking on Brain Energy Metabolism
5.1. Glucose Utilization
5.2. Mitochondrial Function and Oxidative Stress
6. Effects of Diabetes on Brain Energy Metabolism
6.1. Glucose Utilization
6.2. Mitochondrial Function and Oxidative Stress
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Dienel, G.A.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Energy Metabolism of the Brain. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 200–231. [Google Scholar]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-supply of the Brain. J. Physiol. 1890, 11, 85–158.17. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216–216. [Google Scholar] [CrossRef]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front. Neurosci. 2019, 13, 1452. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. The young-adult and normally aged brain. Its blood flow and oxidative metabolism. A review—part I. Arch. Gerontol. Geriatr. 1982, 1, 101–116. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsu, F.M.; Lee, L.W.; Liao, C.L. Energy metabolism during brain ischemia. Stability during reversible and irreversible damage. Stroke 1975, 6, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Writing Group, M.; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–360. [Google Scholar] [CrossRef]
- Sifat, A.E.; Vaidya, B.; Villalba, H.; Albekairi, T.H.; Abbruscato, T.J. Neurovascular unit transport responses to ischemia and common coexisting conditions: Smoking and diabetes. Am. J. Physiol. Cell Physiol. 2019, 316, C2–C15. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Silver, I.A.; Deas, J.; Erecinska, M. Ion homeostasis in brain cells: Differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 1997, 78, 589–601. [Google Scholar] [CrossRef]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Alberg, A.J.; Shopland, D.R.; Cummings, K.M. The 2014 Surgeon General’s report: Commemorating the 50th Anniversary of the 1964 Report of the Advisory Committee to the US Surgeon General and updating the evidence on the health consequences of cigarette smoking. Am. J. Epidemiol. 2014, 179, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Abbruscato, T.J.; Lopez, S.P.; Roder, K.; Paulson, J.R. Regulation of blood-brain barrier Na,K,2Cl-cotransporter through phosphorylation during in vitro stroke conditions and nicotine exposure. J. Pharmacol. Exp. Ther. 2004, 310, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.R.; Yang, T.; Selvaraj, P.K.; Mdzinarishvili, A.; Van der Schyf, C.J.; Klein, J.; Bickel, U.; Abbruscato, T.J. Nicotine exacerbates brain edema during in vitro and in vivo focal ischemic conditions. J. Pharmacol. Exp. Ther. 2010, 332, 371–379. [Google Scholar] [CrossRef]
- Kesarwani, M.; Perez, A.; Lopez, V.A.; Wong, N.D.; Franklin, S.S. Cardiovascular comorbidities and blood pressure control in stroke survivors. J. Hypertens. 2009, 27, 1056–1063. [Google Scholar] [CrossRef]
- Li, P.A.; Siesjo, B.K. Role of hyperglycaemia-related acidosis in ischaemic brain damage. Acta Physiol. Scand. 1997, 161, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, M.; Fujioka, M.; He, Q.; Han, A.; Yong, G.; Csiszar, K.; Li, P.A. Diabetes activates cell death pathway after transient focal cerebral ischemia. Diabetes 2003, 52, 481–486. [Google Scholar] [CrossRef] [Green Version]
- London, E.D.; Connolly, R.J.; Szikszay, M.; Wamsley, J.K.; Dam, M. Effects of nicotine on local cerebral glucose utilization in the rat. J. Neurosci. 1988, 8, 3920–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenco, T.; Bernstein, S.; Cumming, P.; Clarke, P.B. Effects of nicotine and chlorisondamine on cerebral glucose utilization in immobilized and freely-moving rats. Br. J. Pharmacol. 2000, 129, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, J.M.; Gilson, S.F.; Wong, D.F.; Villemagne, V.L.; Dannals, R.F.; Grayson, R.F.; Henningfield, J.E.; London, E.D. Intravenous Nicotine Reduces Cerebral Glucose Metabolism: A Preliminary Study. Neuropsychopharmacology 2003, 28, 765–772. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, L.; Jiang, Y.; Ma, X.; Chowdhury, G.M.; Mason, G.F. Regional metabolite levels and turnover in the awake rat brain under the influence of nicotine. J. Neurochem. 2010, 113, 1447–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Sun, H.; Xu, G.; McCarter, K.D.; Li, J.; Mayhan, W.G. Mito-Tempo prevents nicotine-induced exacerbation of ischemic brain damage. J. Appl. Physiol. 2018, 125, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, A.L. Cerebral glucose metabolism in diabetes mellitus. Eur. J. Pharmacol. 2004, 490, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.M.; Gutiérrez-García, A.G. Cognitive impairment in diabetes and poor glucose utilization in the intracellular neural milieu. Med. Hypotheses 2017, 104, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Triguero, D.; Farrell, C.R. Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes 1990, 39, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Espinosa, M.A.; Garcia-Martin, M.L.; Cerdan, S. Role of glial metabolism in diabetic encephalopathy as detected by high resolution 13C NMR. NMR Biomed. 2003, 16, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, J.; Shi, H. Hyperglycemia as a Risk Factor of Ischemic Stroke. J. Drug Metab. Toxicol. 2013, 4, 153. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.J.; Albers, R.W. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Raven Press: New York, NY, USA, 1994. [Google Scholar]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Bouzier-Sore, A.K.; Merle, M.; Magistretti, P.J.; Pellerin, L. Feeding active neurons: (re)emergence of a nursing role for astrocytes. J. Physiol. Paris 2002, 96, 273–282. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Nissen, J.D.; Waagepetersen, H.S. Improved cerebral energetics and ketone body metabolism in db/db mice. J. Cereb. Blood Flow Metab. 2017, 37, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; d’Avignon, D.A.; Wentz, A.E.; Weber, M.L.; Crawford, P.A. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J. Biol. Chem. 2011, 286, 6902–6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wieman, H.L.; Jacobs, S.R.; Rathmell, J.C. Mechanisms and methods in glucose metabolism and cell death. Meth. Enzymol. 2008, 442, 439–457. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Rapoport, I.; Elsner, R.; Muller, M.; Dumdey, R.; Rapoport, S. NADPH production in the oxidative pentose phosphate pathway as source of reducing equivalents in glycolysis of human red cells in vitro. Acta Biol. Med. Ger. 1979, 38, 901–908. [Google Scholar]
- Winkler, B.S.; DeSantis, N.; Solomon, F. Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Exp. Eye Res. 1986, 43, 829–847. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.J.; Lee, H.; Hong, S.H.; Park, C.; Park, S.H.; Kim, G.Y.; Kim, S.; Kim, H.S.; Hwang, H.J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants (Basel) 2019, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimelberg, H.K.; Norenberg, M.D. Astrocytes. Sci. Am. 1989, 260, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L. Food for thought: The importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. 2010, 36 (Suppl. S3), S59–S63. [Google Scholar] [CrossRef]
- Steele, M.L.; Robinson, S.R. Reactive astrocytes give neurons less support: Implications for Alzheimer’s disease. Neurobiol. Aging 2012, 33, 423.e1–423.e13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef]
- Hertz, L.; Dringen, R.; Schousboe, A.; Robinson, S.R. Astrocytes: Glutamate producers for neurons. J. Neurosci. Res. 1999, 57, 417–428. [Google Scholar] [CrossRef]
- Gjedde, A.; Marrett, S.; Vafaee, M. Oxidative and nonoxidative metabolism of excited neurons and astrocytes. J. Cereb. Blood Flow Metab. 2002, 22, 1–14. [Google Scholar] [CrossRef]
- Cater, H.L.; Chandratheva, A.; Benham, C.D.; Morrison, B., 3rd; Sundstrom, L.E. Lactate and glucose as energy substrates during, and after, oxygen deprivation in rat hippocampal acute and cultured slices. J. Neurochem. 2003, 87, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Cater, H.L.; Benham, C.D.; Sundstrom, L.E. Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J. Physiol. 2001, 531, 459–466. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Pellerin, L. Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann. N. Y. Acad. Sci. 1996, 777, 380–387. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Pellerin, L. Astrocytes Couple Synaptic Activity to Glucose Utilization in the Brain. News Physiol. Sci. 1999, 14, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, R.; Magistretti, P.J.; Pellerin, L. Trans-inhibition of glutamate transport prevents excitatory amino acid-induced glycolysis in astrocytes. Brain Res. 1999, 850, 39–46. [Google Scholar] [CrossRef]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Angamo, E.A.; Rosner, J.; Liotta, A.; Kovacs, R.; Heinemann, U. A neuronal lactate uptake inhibitor slows recovery of extracellular ion concentration changes in the hippocampal CA3 region by affecting energy metabolism. J. Neurophysiol. 2016, 116, 2420–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Esaki, T.; Shimoji, K.; Cook, M.; Law, M.J.; Kaufman, E.; Sokoloff, L. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4879–4884. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Smith, D.; Pernet, A.; Hallett, W.A.; Bingham, E.; Marsden, P.K.; Amiel, S.A. Lactate: A preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 658–664. [Google Scholar] [CrossRef] [Green Version]
- van Hall, G.; Stromstad, M.; Rasmussen, P.; Jans, O.; Zaar, M.; Gam, C.; Quistorff, B.; Secher, N.H.; Nielsen, H.B. Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 2009, 29, 1121–1129. [Google Scholar] [CrossRef]
- Back, T.; Hemmen, T.; Schuler, O.G. Lesion evolution in cerebral ischemia. J. Neurol. 2004, 251, 388–397. [Google Scholar] [CrossRef] [PubMed]
- McCall, A.L.; Van Bueren, A.M.; Nipper, V.; Moholt-Siebert, M.; Downes, H.; Lessov, N. Forebrain ischemia increases GLUT1 protein in brain microvessels and parenchyma. J. Cereb. Blood Flow Metab. 1996, 16, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Vannucci, S.J.; Seaman, L.B.; Vannucci, R.C. Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J. Cereb. Blood Flow Metab. 1996, 16, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gido, G.; Kristian, T.; Siesjo, B.K. Extracellular potassium in a neocortical core area after transient focal ischemia. Stroke 1997, 28, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Kristian, T.; Gido, G.; Kuroda, S.; Schutz, A.; Siesjo, B.K. Calcium metabolism of focal and penumbral tissues in rats subjected to transient middle cerebral artery occlusion. Exp. Brain Res. 1998, 120, 503–509. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.J.; Sharp, F.R.; Heiss, W.D.; Albers, G.W. Heterogeneity in the penumbra. J. Cereb. Blood Flow Metab. 2011, 31, 1836–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folbergrova, J.; Memezawa, H.; Smith, M.L.; Siesjo, B.K. Focal and perifocal changes in tissue energy state during middle cerebral artery occlusion in normo- and hyperglycemic rats. J. Cereb. Blood Flow Metab. 1992, 12, 25–33. [Google Scholar] [CrossRef]
- Paschen, W.; Olah, L.; Mies, G. Effect of transient focal ischemia of mouse brain on energy state and NAD levels: No evidence that NAD depletion plays a major role in secondary disturbances of energy metabolism. J. Neurochem. 2000, 75, 1675–1680. [Google Scholar] [CrossRef]
- Onodera, H.; Iijima, K.; Kogure, K. Mononucleotide metabolism in the rat brain after transient ischemia. J. Neurochem. 1986, 46, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Katsura, K.; Hillered, L.; Bates, T.E.; Siesjo, B.K. Delayed treatment with alpha-phenyl-N-tert-butyl nitrone (PBN) attenuates secondary mitochondrial dysfunction after transient focal cerebral ischemia in the rat. Neurobiol. Dis. 1996, 3, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Kuroda, S.; Kristian, T.; Siesjo, B.K. The immunosuppressant drug FK506 ameliorates secondary mitochondrial dysfunction following transient focal cerebral ischemia in the rat. Neurobiol. Dis. 1997, 4, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Lust, W.D.; Taylor, C.; Pundik, S.; Selman, W.R.; Ratcheson, R.A. Ischemic cell death: Dynamics of delayed secondary energy failure during reperfusion following focal ischemia. Metab. Brain Dis. 2002, 17, 113–121. [Google Scholar] [CrossRef]
- Selman, W.R.; Lust, W.D.; Pundik, S.; Zhou, Y.; Ratcheson, R.A. Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res. 2004, 999, 167–174. [Google Scholar] [CrossRef]
- Lee, D.R.; Helps, S.C.; Macardle, P.J.; Nilsson, M.; Sims, N.R. Alterations in membrane potential in mitochondria isolated from brain subregions during focal cerebral ischemia and early reperfusion: Evaluation using flow cytometry. Neurochem. Res. 2009, 34, 1857–1866. [Google Scholar] [CrossRef]
- Solenski, N.J.; diPierro, C.G.; Trimmer, P.A.; Kwan, A.L.; Helm, G.A. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke 2002, 33, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Carlucci, A.; Adornetto, A.; Scorziello, A.; Viggiano, D.; Foca, M.; Cuomo, O.; Annunziato, L.; Gottesman, M.; Feliciello, A. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008, 27, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korde, A.S.; Pettigrew, L.C.; Craddock, S.D.; Maragos, W.F. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J. Neurochem. 2005, 94, 1676–1684. [Google Scholar] [CrossRef]
- Anderson, M.F.; Sims, N.R. The effects of focal ischemia and reperfusion on the glutathione content of mitochondria from rat brain subregions. J. Neurochem. 2002, 81, 541–549. [Google Scholar] [CrossRef]
- Muyderman, H.; Nilsson, M.; Sims, N.R. Highly selective and prolonged depletion of mitochondrial glutathione in astrocytes markedly increases sensitivity to peroxynitrite. J. Neurosci. 2004, 24, 8019–8028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muyderman, H.; Wadey, A.L.; Nilsson, M.; Sims, N.R. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J. Neurochem. 2007, 102, 1369–1382. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Talley Watts, L.; Holstein, D.M.; Wewer, J.; Lechleiter, J.D. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. J. Cereb. Blood Flow Metab. 2013, 33, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol. 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U.; et al. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef]
- Bath, P.M.; Gray, L.J.; Bath, A.J.; Buchan, A.; Miyata, T.; Green, A.R.; Investigators, N.X.Y.E.M.-a.i.I.A.w.S. Effects of NXY-059 in experimental stroke: An individual animal meta-analysis. Br. J. Pharmacol. 2009, 157, 1157–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, G.F.; Maroz, A.; Cocheme, H.M.; Logan, A.; Prime, T.A.; Peskin, A.V.; Winterbourn, C.C.; James, A.M.; Ross, M.F.; Brooker, S.; et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chem. Biol. 2012, 19, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.F.; Guo, F.; Cao, Y.Z.; Shi, W.; Xia, Q. Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: Antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci. Ther. 2012, 18, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Leng, B. Cynaropicrin Averts the Oxidative Stress and Neuroinflammation in Ischemic/Reperfusion Injury Through the Modulation of NF-kB. Appl. Biochem. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. SIRT1: New avenues of discovery for disorders of oxidative stress. Expert Opin. Ther. Targets 2012, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shan, X.; Li, L.; Dong, L.; Huang, G.; Tao, H. circHIPK3 regulates apoptosis and mitochondrial dysfunction induced by ischemic stroke in mice by sponging miR-148b-3p via CDK5R1/SIRT1. Exp. Neurol. 2022, 355, 114115. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Du, F.; Shih, Y.Y.; Shen, Q.; Gonzalez-Lima, F.; Duong, T.Q. Methylene blue potentiates stimulus-evoked fMRI responses and cerebral oxygen consumption during normoxia and hypoxia. Neuroimage 2013, 72, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Zhang, L.M.; Lu, S.D.; Li, X.J.; Sun, F.Y. Protective effect of melatonin on injuried cerebral neurons is associated with bcl-2 protein over-expression. Acta Pharmacol. Sin. 1999, 20, 409–414. [Google Scholar]
- Cuzzocrea, S.; Costantino, G.; Gitto, E.; Mazzon, E.; Fulia, F.; Serraino, I.; Cordaro, S.; Barberi, I.; De Sarro, A.; Caputi, A.P. Protective effects of melatonin in ischemic brain injury. J. Pineal. Res. 2000, 29, 217–227. [Google Scholar] [CrossRef]
- Zheng, Y.; Hou, J.; Liu, J.; Yao, M.; Li, L.; Zhang, B.; Zhu, H.; Wang, Z. Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. J. Pharmacol. Sci. 2014, 124, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Kilic, U.; Elibol, B.; Caglayan, A.B.; Beker, M.C.; Beker, M.; Altug-Tasa, B.; Uysal, O.; Yilmaz, B.; Kilic, E. Delayed Therapeutic Administration of Melatonin Enhances Neuronal Survival Through AKT and MAPK Signaling Pathways Following Focal Brain Ischemia in Mice. J. Mol. Neurosci. 2022, 72, 994–1007. [Google Scholar] [CrossRef]
- Tay, A.S.; Hu, L.F.; Lu, M.; Wong, P.T.; Bian, J.S. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway. Neuroscience 2010, 167, 277–286. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents OGD/R-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831. [Google Scholar] [CrossRef]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain. Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.; Choteau, F.; Pucci, B.; Villamena, F.A. Reactivity of superoxide radical anion and hydroperoxyl radical with alpha-phenyl-N-tert-butylnitrone (PBN) derivatives. J. Phys. Chem. A 2008, 112, 12498–12509. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Puchowicz, M.A.; Sun, X.; LaManna, J.C. Mitochondrial dysfunction in aging rat brain following transient global ischemia. Adv. Exp. Med. Biol. 2008, 614, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, M.; Lennmyr, F. Neuroprotection by S-PBN in hyperglycemic ischemic brain injury in rats. Ups. J. Med. Sci. 2010, 115, 163–168. [Google Scholar] [CrossRef]
- Zhao, G.; Yao-Yue, C.; Qin, G.W.; Guo, L.H. Luteolin from Purple Perilla mitigates ROS insult particularly in primary neurons. Neurobiol. Aging 2012, 33, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Zang, S.Y.; Jiang, Z.H.; Chen, Y.Y.; Ji, X.H.; Lu, B.F.; Wu, J.H.; Qin, G.W.; Guo, L.H. Postischemic administration of liposome-encapsulated luteolin prevents against ischemia-reperfusion injury in a rat middle cerebral artery occlusion model. J. Nutr. Biochem. 2011, 22, 929–936. [Google Scholar] [CrossRef]
- Qiao, H.; Dong, L.; Zhang, X.; Zhu, C.; Zhang, X.; Wang, L.; Liu, Z.; Chen, L.; Xing, Y.; Wang, C.; et al. Protective effect of luteolin in experimental ischemic stroke: Upregulated SOD1, CAT, Bcl-2 and claudin-5, down-regulated MDA and Bax expression. Neurochem. Res. 2012, 37, 2014–2024. [Google Scholar] [CrossRef]
- Dong, R.; Huang, R.; Shi, X.; Xu, Z.; Mang, J. Exploration of the mechanism of luteolin against ischemic stroke based on network pharmacology, molecular docking and experimental verification. Bioengineered 2021, 12, 12274–12293. [Google Scholar] [CrossRef] [PubMed]
- Bruning, C.A.; Prigol, M.; Luchese, C.; Jesse, C.R.; Duarte, M.M.; Roman, S.S.; Nogueira, C.W. Protective effect of diphenyl diselenide on ischemia and reperfusion-induced cerebral injury: Involvement of oxidative stress and pro-inflammatory cytokines. Neurochem. Res. 2012, 37, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Dobrachinski, F.; da Silva, M.H.; Tassi, C.L.; de Carvalho, N.R.; Dias, G.R.; Golombieski, R.M.; da Silva Loreto, E.L.; da Rocha, J.B.; Fighera, M.R.; Soares, F.A. Neuroprotective effect of diphenyl diselenide in a experimental stroke model: Maintenance of redox system in mitochondria of brain regions. Neurotox. Res. 2014, 26, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, Y.; Ma, Y.; Zhang, X.; Yang, L.; Shen, X.; Zhang, J.; Jing, L. Selenium attenuates ischemia/reperfusion injuryinduced damage to the bloodbrain barrier in hyperglycemia through PI3K/AKT/mTOR pathwaymediated autophagy inhibition. Int. J. Mol. Med. 2021, 48. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Miller, A.A. Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy? Front. Pharmacol. 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayarani, I.; Chaudiere, J.; Lefauconnier, J.M.; Bourre, J.M. Enzymatic protection against peroxidative damage in isolated brain capillaries. J. Neurochem. 1987, 48, 1399–1402. [Google Scholar] [CrossRef]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging 2001, 18, 685–716. [Google Scholar] [CrossRef]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enciu, A.M.; Gherghiceanu, M.; Popescu, B.O. Triggers and effectors of oxidative stress at blood-brain barrier level: Relevance for brain ageing and neurodegeneration. Oxid. Med. Cell Longev. 2013, 2013, 297512. [Google Scholar] [CrossRef]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Shah, G.N.; Morofuji, Y.; Banks, W.A.; Price, T.O. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: Implications for cerebral microvascular disease in diabetes. Biochem. Biophys. Res. Commun. 2013, 440, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, M.; Gu, R.; Xu, G.; Wu, H. Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 777–788. [Google Scholar] [CrossRef]
- Kontos, C.D.; Wei, E.P.; Williams, J.I.; Kontos, H.A.; Povlishock, J.T. Cytochemical detection of superoxide in cerebral inflammation and ischemia in vivo. Am. J. Physiol. 1992, 263, H1234–1242. [Google Scholar] [CrossRef]
- Liu, T.H.; Beckman, J.S.; Freeman, B.A.; Hogan, E.L.; Hsu, C.Y. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am. J. Physiol. 1989, 256, H589–593. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.J.; Shin, D.H.; Im, D.S.; Bang, O.Y.; Nam, H.S.; Lee, J.H.; Joo, I.S.; Huh, K.; Gwag, B.J. Identification of oxidized serum albumin in the cerebrospinal fluid of ischaemic stroke patients. Eur. J. Neurol. 2011, 18, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.; Back, T.; Lindauer, U.; Busch, C.; Megow, D.; Dreier, J.; Dirnagl, U. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J. Cereb. Blood Flow Metab. 1998, 18, 196–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A.; Zhang, J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 1996, 27, 327–331, discussion 332. [Google Scholar] [CrossRef] [PubMed]
- Cino, M.; Del Maestro, R.F. Generation of hydrogen peroxide by brain mitochondria: The effect of reoxygenation following postdecapitative ischemia. Arch. Biochem. Biophys. 1989, 269, 623–638. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhong, W.; Tu, Q.; Ding, B. NADPH oxidase mediates the expression of MMP-9 in cerebral tissue after ischemia-reperfusion damage. Neurol. Res. 2014, 36, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Weston, R.M.; Dusting, G.J.; Roulston, C.L. NADPH oxidase and angiogenesis following endothelin-1 induced stroke in rats: Role for nox2 in brain repair. Brain Sci. 2013, 3, 294–317. [Google Scholar] [CrossRef]
- Patt, A.; Harken, A.H.; Burton, L.K.; Rodell, T.C.; Piermattei, D.; Schorr, W.J.; Parker, N.B.; Berger, E.M.; Horesh, I.R.; Terada, L.S.; et al. Xanthine oxidase-derived hydrogen peroxide contributes to ischemia reperfusion-induced edema in gerbil brains. J. Clin. Investig. 1988, 81, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, T.; Tsuruta, R.; Fujita, M.; Aki, H.S.; Kutsuna, S.; Kawamura, Y.; Wakatsuki, J.; Aoki, T.; Kobayashi, C.; Kasaoka, S.; et al. Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res. 2009, 1305, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstead, W.M.; Mirro, R.; Busija, D.W.; Leffler, C.W. Postischemic generation of superoxide anion by newborn pig brain. Am. J. Physiol. 1988, 255, H401–403. [Google Scholar] [CrossRef] [PubMed]
- Mizuma, A.; Yenari, M.A. Anti-Inflammatory Targets for the Treatment of Reperfusion Injury in Stroke. Front. Neurol. 2017, 8, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [Green Version]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 2020, 11, 1559. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Shah, R.S.; Cole, J.W. Smoking and stroke: The more you smoke the more you stroke. Expert Rev. Cardiovasc. Ther. 2010, 8, 917–932. [Google Scholar] [CrossRef]
- Bonita, R.; Duncan, J.; Truelsen, T.; Jackson, R.T.; Beaglehole, R. Passive smoking as well as active smoking increases the risk of acute stroke. Tob. Control 1999, 8, 156–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Diabetes. Available online: https://www.cdc.gov/diabetes/basics/diabetes.html (accessed on 13 March 2021).
- Towfighi, A.; Markovic, D.; Ovbiagele, B. Current national patterns of comorbid diabetes among acute ischemic stroke patients. Cerebrovasc. Dis. 2012, 33, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Maida, C.; Maugeri, R.; Iacopino, G.; Pinto, A. Relationship between diabetes and ischemic stroke: Analysis of diabetes-related risk factors for stroke and of specific patterns of stroke associated with diabetes mellitus. J. Diabetes Metab. 2015, 6, 544–551. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Katsanos, A.H.; Mavridis, D.; Lambadiari, V.; Roffe, C.; Macleod, M.J.; Sevcik, P.; Cappellari, M.; Nevsimalova, M.; Toni, D.; et al. Association of Baseline Hyperglycemia with Outcomes of Patients with and without Diabetes with Acute Ischemic Stroke Treated with Intravenous Thrombolysis: A Propensity Score-Matched Analysis from the SITS-ISTR Registry. Diabetes 2019, 68, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.B.; Kim, D.K.; Park, D.J.; Shah, M.A.; Kim, M.O.; Jung, E.J.; Lee, H.S.; Koh, P.O. Hyperglycemia aggravates decrease in alpha-synuclein expression in a middle cerebral artery occlusion model. Lab. Anim. Res. 2018, 34, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Desilles, J.P.; Syvannarath, V.; Ollivier, V.; Journe, C.; Delbosc, S.; Ducroux, C.; Boisseau, W.; Louedec, L.; Di Meglio, L.; Loyau, S.; et al. Exacerbation of Thromboinflammation by Hyperglycemia Precipitates Cerebral Infarct Growth and Hemorrhagic Transformation. Stroke 2017, 48, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Portier, I.; Kosaka, Y.; Campbell, R.A. Hyperglycemia exacerbates ischemic stroke outcome independent of platelet glucose uptake. J. Thromb. Haemost. 2021, 19, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.K.; Boreddy, P.R.; Abbruscato, T.J. Nicotine pre-exposure reduces stroke-induced glucose transporter-1 activity at the blood-brain barrier in mice. Fluids Barriers CNS 2015, 12, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasu, C. Glycoxidative stress and cardiovascular complications in experimentally-induced diabetes: Effects of antioxidant treatment. Open Cardiovasc. Med. J. 2010, 4, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünwald, F.; Schröck, H.; Kuschinsky, W. The effect of an acute nicotine infusion on the local cerebral glucose utilization of the awake rat. Brain Res. 1987, 400, 232–238. [Google Scholar] [CrossRef]
- Domino, E.F.; Minoshima, S.; Guthrie, S.K.; Ohl, L.; Ni, L.; Koeppe, R.A.; Cross, D.J.; Zubieta, J.K. Effects of nicotine on regional cerebral glucose metabolism in awake resting tobacco smokers. Neuroscience 2000, 101, 277–282. [Google Scholar] [CrossRef]
- Vafaee, M.S.; Gjedde, A.; Imamirad, N.; Vang, K.; Chakravarty, M.M.; Lerch, J.P.; Cumming, P. Smoking normalizes cerebral blood flow and oxygen consumption after 12-hour abstention. J. Cereb. Blood Flow Metab. 2015, 35, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Goniewicz, M.L.; Kuma, T.; Gawron, M.; Knysak, J.; Kosmider, L. Nicotine levels in electronic cigarettes. Nicotine Tob. Res. 2013, 15, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benowitz, N.L.; Hukkanen, J.; Jacob, P., 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb. Exp. Pharmacol. 2009, 29–60. [Google Scholar] [CrossRef] [Green Version]
- Chase, H.P.; Garg, S.K.; Marshall, G.; Berg, C.L.; Harris, S.; Jackson, W.E.; Hamman, R.E. Cigarette Smoking Increases the Risk of Albuminuria among Subjects with Type I Diabetes. JAMA 1991, 265, 614–617. [Google Scholar] [CrossRef]
- Chiolero, A.; Faeh, D.; Paccaud, F.; Cornuz, J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am. J. Clin. Nutr. 2008, 87, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsbad, S.; McNair, P.; Christensen, M.S.; Christiansen, C.; Faber, O.K.; Binder, C.; Transbøl, I. Influence of Smoking on Insulin Requirement and Metbolic Status in Diabetes Mellitus. Diabetes Care 1980, 3, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Will, J.C.; Galuska, D.A.; Ford, E.S.; Mokdad, A.; Calle, E.E. Cigarette smoking and diabetes mellitus: Evidence of a positive association from a large prospective cohort study. Int. J. Epidemiol. 2001, 30, 540–546. [Google Scholar] [CrossRef] [Green Version]
- Sifat, A.E.; Nozohouri, S.; Villalba, H.; Al Shoyaib, A.; Vaidya, B.; Karamyan, V.T.; Abbruscato, T. Prenatal electronic cigarette exposure decreases brain glucose utilization and worsens outcome in offspring hypoxic-ischemic brain injury. J. Neurochem. 2020, 153, 63–79. [Google Scholar] [CrossRef]
- Sifat, A.E.; Vaidya, B.; Kaisar, M.A.; Cucullo, L.; Abbruscato, T.J. Nicotine and electronic cigarette (E-Cig) exposure decreases brain glucose utilization in ischemic stroke. J. Neurochem. 2018, 147, 204–221. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Raval, A.P. Simultaneous nicotine and oral contraceptive exposure alters brain energy metabolism and exacerbates ischemic stroke injury in female rats. J. Cereb. Blood Flow Metab. 2021, 41, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Shen, F.; He, Y.; Degos, V.; Camus, M.; Maze, M.; Young, W.L.; Su, H. Activation of alpha-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS ONE 2014, 9, e105711. [Google Scholar] [CrossRef]
- Zou, D.; Luo, M.; Han, Z.; Zhan, L.; Zhu, W.; Kang, S.; Bao, C.; Li, Z.; Nelson, J.; Zhang, R.; et al. Activation of Alpha-7 Nicotinic Acetylcholine Receptor Reduces Brain Edema in Mice with Ischemic Stroke and Bone Fracture. Mol. Neurobiol. 2017, 54, 8278–8286. [Google Scholar] [CrossRef] [PubMed]
- Malinska, D.; Wieckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymanski, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.P.; Lagrue, G. In vitro effects of nicotine on mitochondrial respiration and superoxide anion generation. Brain Res. 2001, 900, 72–79. [Google Scholar] [CrossRef]
- Guo, L.; Li, L.; Wang, W.; Pan, Z.; Zhou, Q.; Wu, Z. Mitochondrial reactive oxygen species mediates nicotine-induced hypoxia-inducible factor-1alpha expression in human non-small cell lung cancer cells. Biochim. Biophys. Acta 2012, 1822, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kim, J.-M.; Donovan, D.M.; Becker, K.G.; Li, M.D. Significant modulation of mitochondrial electron transport system by nicotine in various rat brain regions. Mitochondrion 2009, 9, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Raval, A.P.; Dave, K.R.; Saul, I.; Gonzalez, G.J.; Diaz, F. Synergistic inhibitory effect of nicotine plus oral contraceptive on mitochondrial complex-IV is mediated by estrogen receptor-β in female rats. J. Neurochem. 2012, 121, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Lerner, C.; Sundar, I.K.; Rahman, I. Myofibroblast differentiation and its functional properties are inhibited by nicotine and e-cigarette via mitochondrial OXPHOS complex III. Sci. Rep. 2017, 7, 43213. [Google Scholar] [CrossRef] [Green Version]
- Sachs, C.; Jonsson, G. Mechanisms of action of 6-hydroxydopamine. Biochem. Pharmacol. 1975, 24, 1–8. [Google Scholar] [CrossRef]
- Perumal, A.S.; Gopal, V.B.; Tordzro, W.K.; Cooper, T.B.; Cadet, J.L. Vitamin E attenuates the toxic effects of 6-hydroxydopamine on free radical scavenging systems in rat brain. Brain Res. Bull. 1992, 29, 699–701. [Google Scholar] [CrossRef]
- Kumar, R.; Agarwal, A.K.; Seth, P.K. Free radical-generated neurotoxicity of 6-hydroxydopamine. J. Neurochem. 1995, 64, 1703–1707. [Google Scholar] [CrossRef]
- Hritcu, L.; Ciobica, A.; Gorgan, L. Nicotine-induced memory impairment by increasing brain oxidative stress. Cent. Eur. J. Biol. 2009, 4, 335–342. [Google Scholar] [CrossRef]
- Barros, D.M.; Galhardi, F.G.; Ferreira, J.R.; Guterres, L.B.; Dickel, O.; Geracitano, L.; Izquierdo, I.; Monserrat, J.M. The benefits and drawbacks of nicotine exposure in the cortex and hippocampus of old rats. Neurotoxicology 2007, 28, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Gautam, N.; Dey, S.K.; Maiti, T.; Roy, S. Oxidative stress in the brain of nicotine-induced toxicity: Protective role of Andrographis paniculata Nees and vitamin E. Appl. Physiol. Nutr. Metab. 2009, 34, 124–135. [Google Scholar] [CrossRef]
- Ande, A.; Earla, R.; Jin, M.; Silverstein, P.S.; Mitra, A.K.; Kumar, A.; Kumar, S. An LC–MS/MS method for concurrent determination of nicotine metabolites and the role of CYP2A6 in nicotine metabolite-mediated oxidative stress in SVGA astrocytes. Drug Alcohol Depend. 2012, 125, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Dillip, K.M.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef]
- Sussan, T.E.; Gajghate, S.; Thimmulappa, R.K.; Ma, J.; Kim, J.H.; Sudini, K.; Consolini, N.; Cormier, S.A.; Lomnicki, S.; Hasan, F.; et al. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS ONE 2015, 10, e0116861. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S.; Morita, T.; Liu, Y.; Christou, H. Mechanisms by which oxygen regulates gene expression and cell-cell interaction in the vasculature. Kidney Int. 1997, 51, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael Pittilo, R. Cigarette smoking, endothelial injury and cardiovascular disease. Int. J. Exp. Pathol. 2000, 81, 219–230. [Google Scholar] [CrossRef]
- Pittilo, R.; Woolf, N. Cigarette smoking as a risk factor for atherosclerosis. J. Smoking-Related. Dis. 1994, 5, 43–47. [Google Scholar]
- Shimosato, T.; Geddawy, A.; Tawa, M.; Imamura, T.; Okamura, T. Chronic administration of nicotine-free cigarette smoke extract impaired endothelium-dependent vascular relaxation in rats via increased vascular oxidative stress. J. Pharmacol. Sci. 2012, 118, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, P.; Fofaria, N.; Prasad, S.; Sajja, R.K.; Weksler, B.; Couraud, P.O.; Romero, I.A.; Cucullo, L. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: Is smoking reduced or nicotine-free products really safe? BMC Neurosci. 2014, 15, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, S.L.; Lannan, K.L.; Loelius, S.G.; Phipps, R.P. In Vitro and Ex Vivo Approaches to Evaluate Next-Generation Tobacco and Non-Tobacco Products on Human Blood Platelets. Appl. In Vitro Toxicol. 2017, 3, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sithu, S.D.; Srivastava, S.; Siddiqui, M.A.; Vladykovskaya, E.; Riggs, D.W.; Conklin, D.J.; Haberzettl, P.; O’Toole, T.E.; Bhatnagar, A.; D’Souza, S.E. Exposure to acrolein by inhalation causes platelet activation. Toxicol. Appl. Pharmacol. 2010, 248, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, R.B.; Kraal, J.H.; Huang, L.J.; Chancellor, M.B. Effects of cigarette smoke components on in vitro chemotaxis of human polymorphonuclear leukocytes. Infect. Immun. 1977, 16, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappacosta, B.; Persichilli, S.; Minucci, A.; Stasio, E.D.; Carlino, P.; Pagliari, G.; Giardina, B.; Sole, P.D. Effect of aqueous cigarette smoke extract on the chemiluminescence kinetics of polymorphonuclear leukocytes and on their glycolytic and phagocytic activity. Luminescence 2001, 16, 315–319. [Google Scholar] [CrossRef]
- Lee, H.L.; Chen, C.L.; Yeh, S.T.; Zweier, J.L.; Chen, Y.R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Sun, H.; Arrick, D.M.; Mayhan, W.G. Chronic nicotine exposure exacerbates transient focal cerebral ischemia-induced brain injury. J. Appl. Physiol. 2016, 120, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.P.; Lagrue, G. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology 2003, 44, 642–652. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef]
- Premilovac, D.; Gasperini, R.J.; Sawyer, S.; West, A.; Keske, M.A.; Taylor, B.V.; Foa, L. A New Method for Targeted and Sustained Induction of Type 2 Diabetes in Rodents. Sci. Rep. 2017, 7, 14158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, C.; Zhu, C.; Yu, W.; Jiang, X.; Zhang, F. High-Fat Diet/Low-Dose Streptozotocin-Induced Type 2 Diabetes in Rats Impacts Osteogenesis and Wnt Signaling in Bone Marrow Stromal Cells. PLoS ONE 2015, 10, e0136390. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, A.; Crone, C. Blood-brain glucose transfer: Repression in chronic hyperglycemia. Science 1981, 214, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Reagan, L.P.; Rosell, D.R.; Alves, S.E.; Hoskin, E.K.; McCall, A.L.; Charron, M.J.; McEwen, B.S. GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res. 2002, 932, 129–134. [Google Scholar] [CrossRef]
- Lapidot, A.; Haber, S. Effect of endogenous β-hydroxybutyrate on glucose metabolism in the diabetic rabbit brain: A 13C-magnetic resonance spectroscopy study of [U-13C] glucose metabolites. J. Neurosci. Res. 2001, 64, 207–216. [Google Scholar] [CrossRef]
- Mans, A.; DeJoseph, M.; Davis, D.; Hawkins, R. Brain energy metabolism in streptozotocin-diabetes. Biochem. J. 1988, 249, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Roy Choudhury, G.; Winters, A.; Prah, J.; Lin, W.; Liu, R.; Yang, S.H. Hyperglycemia Alters Astrocyte Metabolism and Inhibits Astrocyte Proliferation. Aging Dis. 2018, 9, 674–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, S.J.; Gibbs, E.M.; Simpson, I.A. Glucose utilization and glucose transporter proteins GLUT-1 and GLUT-3 in brains of diabetic (db/db) mice. Am. J. Physiol. 1997, 272, E267–E274. [Google Scholar] [CrossRef]
- Soares, E.; Prediger, R.D.; Nunes, S.; Castro, A.A.; Viana, S.D.; Lemos, C.; De Souza, C.M.; Agostinho, P.; Cunha, R.A.; Carvalho, E. Spatial memory impairments in a prediabetic rat model. Neuroscience 2013, 250, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Patil, I.; Sancheti, H.; Yin, F.; Cadenas, E. Effects of lipoic acid on high-fat diet-induced alteration of synaptic plasticity and brain glucose metabolism: A PET/CT and 13 C-NMR study. Sci. Rep. 2017, 7, 5391. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef]
- Sickmann, H.M.; Waagepetersen, H.S.; Schousboe, A.; Benie, A.J.; Bouman, S.D. Obesity and type 2 diabetes in rats are associated with altered brain glycogen and amino-acid homeostasis. J. Cereb. Blood Flow Metab. 2010, 30, 1527–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sickmann, H.M.; Waagepetersen, H.S.; Schousboe, A.; Benie, A.J.; Bouman, S.D. Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem. Int. 2012, 60, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Lu, X.; Zhan, L.; Qi, X.; Liang, L.; Hu, S.; Yan, Y.; Zhao, S.; Sui, H.; Zhang, F. The effects of the Chinese medicine ZiBu PiYin recipe on the hippocampus in a rat model of diabetes-associated cognitive decline: A proteomic analysis. Diabetologia 2011, 54, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S.; Lannert, H. Long-term effects of corticosterone on behavior, oxidative and energy metabolism of parietotemporal cerebral cortex and hippocampus of rats: Comparison to intracerebroventricular streptozotocin. J. Neural Transm. (Vienna) 2008, 115, 1241–1249. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seiça, R.; Oliveira, C.R. Increased Vulnerability of Brain Mitochondria in Diabetic (Goto-Kakizaki) Rats with Aging and Amyloid-β Exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2009, 53, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acar, A.; Akil, E.; Alp, H.; Evliyaoglu, O.; Kibrisli, E.; Inal, A.; Unan, F.; Tasdemir, N. Oxidative damage is ameliorated by curcumin treatment in brain and sciatic nerve of diabetic rats. Int. J. Neurosci. 2012, 122, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Aleisa, A.M.; Al-Rejaie, S.S.; Abuohashish, H.M.; Parmar, M.Y.; Alhomida, A.S.; Ahmed, M.M. Flavonoid, morin inhibits oxidative stress, inflammation and enhances neurotrophic support in the brain of streptozotocin-induced diabetic rats. Neurol. Sci. 2014, 35, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Avila, O.; Esquivel-Martinez, M.; Olmos-Orizaba, B.E.; Saavedra-Molina, A.; Rodriguez-Orozco, A.R.; Cortes-Rojo, C. Avocado Oil Improves Mitochondrial Function and Decreases Oxidative Stress in Brain of Diabetic Rats. J. Diabetes Res. 2015, 2015, 485759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan, L.P.; Magarinos, A.M.; McEwen, B.S. Neurological changes induced by stress in streptozotocin diabetic rats. Ann. N. Y. Acad. Sci. 1999, 893, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol. 2005, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Restivo, F.; Vercellinatto, I.; Danni, O.; Brignardello, E.; Aragno, M.; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Tigchelaar, C.; van Zuylen, M.L.; Hulst, A.H.; Preckel, B.; van Beek, A.P.; Kema, I.P.; Hermanides, J.; Absalom, A.R. Elevated cerebrospinal fluid glucose levels and diabetes mellitus are associated with activation of the neurotoxic polyol pathway. Diabetologia 2022, 65, 1098–1107. [Google Scholar] [CrossRef]
- Rajchgot, T.; Thomas, S.C.; Wang, J.C.; Ahmadi, M.; Balood, M.; Crosson, T.; Dias, J.P.; Couture, R.; Claing, A.; Talbot, S. Neurons and Microglia; A Sickly-Sweet Duo in Diabetic Pain Neuropathy. Front. Neurosci. 2019, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Cisneros, R.; Cortés-Rojo, C.; Manzo-Avalos, S.; Clemente-Guerrero, M.; Calderón-Cortés, E.; Salgado-Garciglia, R.; Montoya-Pérez, R.; Boldogh, I.; Saavedra-Molina, A. Mitochondrial response to oxidative and nitrosative stress in early stages of diabetes. Mitochondrion 2013, 13, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Rolo, A.P.; Sena, C.; Seica, R.; Oliveira, C.R.; Santos, M.S. Insulin attenuates diabetes-related mitochondrial alterations: A comparative study. Med. Chem. 2006, 2, 299–308. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Rehni, A.K.; Nautiyal, N.; Perez-Pinzon, M.A.; Dave, K.R. Hyperglycemia/hypoglycemia-induced mitochondrial dysfunction and cerebral ischemic damage in diabetics. Metab. Brain Dis. 2015, 30, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, L.; Dong, Y.; Kumar, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics: An emerging paradigm in ischemia-reperfusion injury. Curr. Pharm. Des. 2013, 19, 6848–6857. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumini-Oliveira, J.; Magalhaes, J.; Pereira, C.V.; Moreira, A.C.; Oliveira, P.J.; Ascensao, A. Endurance training reverts heart mitochondrial dysfunction, permeability transition and apoptotic signaling in long-term severe hyperglycemia. Mitochondrion 2011, 11, 54–63. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, N.E.; Cotter, M.A.; Hohman, T.C. Interactions between essential fatty acid, prostanoid, polyol pathway and nitric oxide mechanisms in the neurovascular deficit of diabetic rats. Diabetologia 1996, 39, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneto, H.; Fujii, J.; Suzuki, K.; Kasai, H.; Kawamori, R.; Kamada, T.; Taniguchi, N. DNA cleavage induced by glycation of Cu,Zn-superoxide dismutase. Biochem. J. 1994, 304 Pt 1, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B., 3rd. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Nozohouri, S.; Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Novel approaches for the delivery of therapeutics in ischemic stroke. Drug Discov. Today 2020, 25, 535–551. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacologic Class | Description/Mechanism of Action(s) | Stroke Model Used | Species and Number of Animals | Sex of Animals | Therapeutic Outcome(s) | Year Published with Reference(s) |
|---|---|---|---|---|---|---|
| Mitochondrial fission inhibitor (Mdivi) | -Inhibition of the assembly of Drp1 and GTPase Drp1 enzymatic activity -Reduction of the level of Drp1 and Cytochrome c | tMCAO | Wistar rats, 20/group | Male | -Reduced cerebral damage induced by ischemia-reperfusion injury -Inhibition of apoptotic cell deaths | 2013 [84], 2014 [85] |
| Modulators of purinergic receptors | -Stimulation of glia-specific purinergic receptor, P2Y1R -Increased mitochondrial O2 consumption and ATP production -P2X7R antagonism decreased expressions of P2X7R, NLRP3, ASC, Caspase-1 p20, and cleaved caspase-3 in ischemic brain tissue | Photothrombotic model | - Transgenic mice -C57BL/6J mice, 3 or 6/group | Male | -Reduced neuronal damage, cell death, and swelling in ischemic stroke -Reduced brain infarct size and neuronal apoptosis -Improved functional outcome after stroke | 2013 [86], 2017 [87] |
| Antioxidants and SOD mimetics | -Free radical trapping -Mitochondria-specific reduction of O2−, cytochorme c, caspase-3, and CHOP -Inhibition of the NF-κB pathway | -pMCAO -tMCAO | C57BL/6J mice; 6/group, Wistar rats, 12/group | Male | -Decresed brain lesion volume, motor impairment, and neglect in animal models -Reduced brain infarct volume, tissue damage, and apoptosis | 2007 [88], 2009 [89], 2012 [90,91], 2022 [92] |
| Activators of NAD-dependent deacetylase sirtuin 1 (SIRT1) | -Reduction of inflammation and oxidative stress -Prevention of lipid peroxidation -Mimicking ischemic preconditioning in brain -Alteration of CDK5R1/SIRT1 signaling | Global cerebral ischemia followed by asphyxial cardiac arrest | Sprague Dawley (SD) rats, 5 or 8/group C57BL/6J mice, 15/group | Male | -Reduced brain infarct volume and neurological deficits -Improved regional brain blood flow, apoptosis, and mitochondrial dysfunctions | 2009 [93], 2012 [94], 2022 [95] |
| Methylene blue | -Alternative electron carrier which reduces electron leakage and ROS production -Enhancing mitochondrial oxygen consumption rate and decreasing the extracellular acidification rate | -tMCAO -Global hypoxia (15% O2) | -Sprague-Dawley rats -Sprague-Dawley rats, 6/group | Male | -Reduced ischemic brain infarct volume | 2011 [96], 2013 [97] |
| Melatonin | -Enhancing the expression of neuronal bcl-2 -Inhibition of autophagy -Activation of the PI3K/Akt pro-survival pathway -Reduction of oxidative stress -Inhibition of MAPK pathway | -tMCAO -BCO | -Rats -Mongolian gerbils, 10/group | Male | -Decreased brain infarct area and neurological impairments -Reduction of post-ischemic brain area -Increased survival and reduced hyperactivity | 1999 [98], 2000 [99], 2014 [100], 2022 [101] |
| Hydrogen sulfide (H2S) | -Stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway -Inhibition of ROS and caspase-3 | Four artery occlusion | Sprague-Dawley rats, 6/group | Male | -Neuroprotection in ischemic neurons -Reduction of neuronal apoptosis | 2010 [102], 2013 [103,104] |
| Alpha-phenyl-tert-butyl-nitrone (PBN) | -Free radical scavenger -Improved mitochondrial respiratory function | -Total cerebral ischemia -tMCAO | -Fischer 344 rats, 3-5/group -Sprague-Dawley rats, 6-7/group | Male | -Improved neurological performance | 2008 [105,106], 2010 [107] |
| Luteolin | -Decrease in ROS production -Protecting the activities of mitochondria, catalase, and glutathione -TNF signaling pathway | -tMCAO -pMCAO | Sprague-Dawley rats, 16-18/group, Sprague-Dawley rats, 6-10/group | -Female -Male | -Reduced brain infarct volume -Improved behavioral and motor functions after stroke | 2011 [108], 2012 [109,110], 2021 [111] |
| Selenium compounds | -Reduction of oxidative stress (ROS, malondialdehyde) and proinflammatory cytokines -Protection of mitochondrial dehydrogenase and complex I activity and reduced mitochondrial swelling -Decreased autophagy | -tBCCAO -tMCAO | Wistar rats, 8/group Diabetic Sprague-Dawley rats, 20/group | Male | -Improved brain infarct and edema -Decreased BBB damage -Improved neurological functions | 2012 [112], 2014 [113], 2021 [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sifat, A.E.; Nozohouri, S.; Archie, S.R.; Chowdhury, E.A.; Abbruscato, T.J. Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes. Int. J. Mol. Sci. 2022, 23, 8512. https://doi.org/10.3390/ijms23158512
Sifat AE, Nozohouri S, Archie SR, Chowdhury EA, Abbruscato TJ. Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes. International Journal of Molecular Sciences. 2022; 23(15):8512. https://doi.org/10.3390/ijms23158512
Chicago/Turabian StyleSifat, Ali Ehsan, Saeideh Nozohouri, Sabrina Rahman Archie, Ekram Ahmed Chowdhury, and Thomas J. Abbruscato. 2022. "Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes" International Journal of Molecular Sciences 23, no. 15: 8512. https://doi.org/10.3390/ijms23158512
APA StyleSifat, A. E., Nozohouri, S., Archie, S. R., Chowdhury, E. A., & Abbruscato, T. J. (2022). Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes. International Journal of Molecular Sciences, 23(15), 8512. https://doi.org/10.3390/ijms23158512