
Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
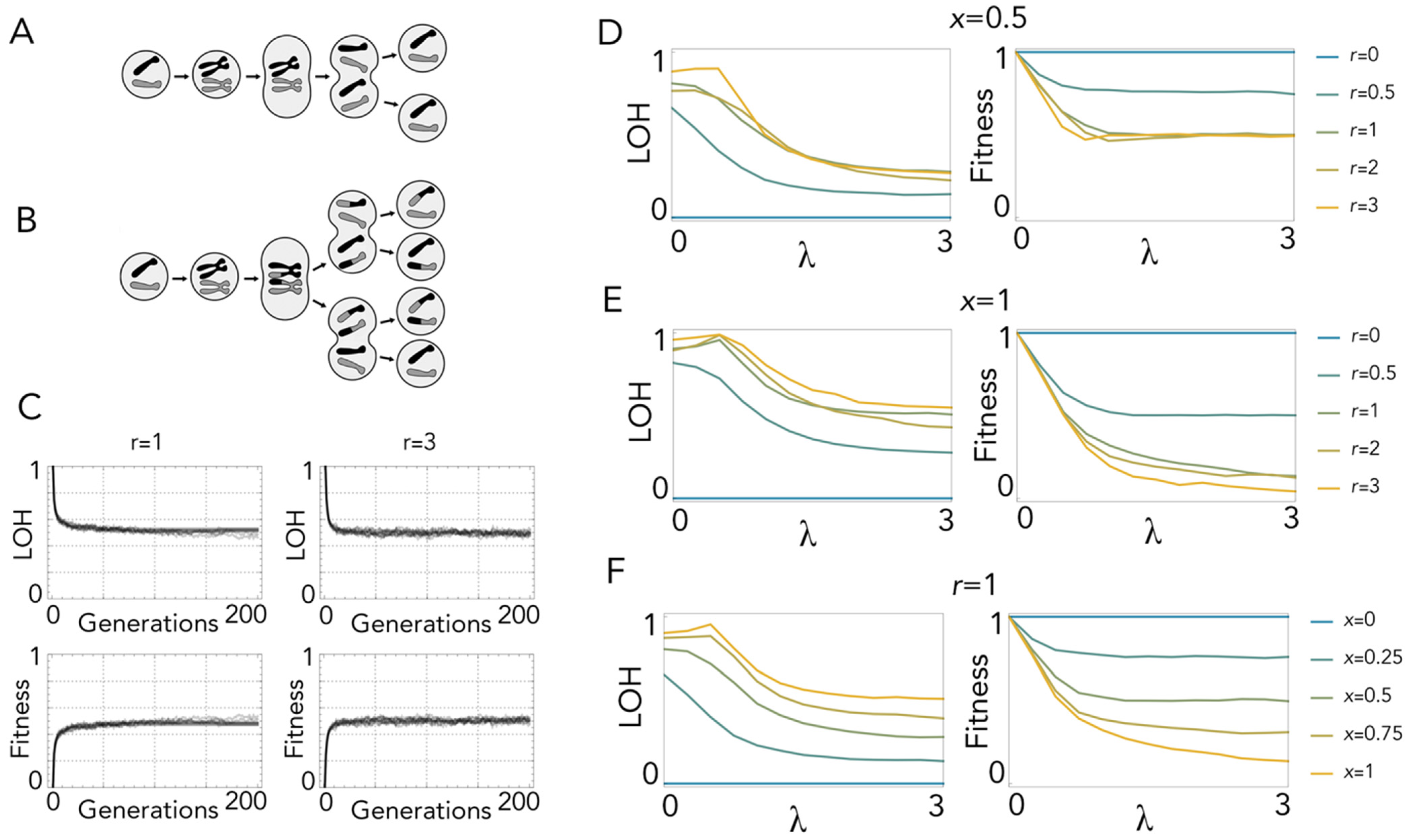
2. Results
2.1. Loss of Heterozygosity in Mitosis with Recombination
2.2. Polyploidy Slows down Loss of Heterozygosity
2.3. Endomitosis Enables a Lower Loss of Heterozygosity Than Mitosis
3. Discussion
4. Methods
- The number of homozygous loci is measured.
- Fitness is calculated as 1-h·λ, where h is the fraction of homozygous loci and λ is the number of lethal equivalents (one lethal equivalent is one recessive mutation whose effect—or more mutations whose summed effects—is lethal when made homozygous); if h·λ > 1, fitness is equal to 0 (because an individual can have more than 1 lethal equivalent but cannot die more than once).
- A cell is chosen to reproduce with a probability proportional to its fitness.
- For endomitosis only: chromosomes are duplicated and paired; pairing occurs between sister chromosomes with probability s or between non-sister chromosomes with probability 1-s.
- If 0 < r < 1, a locus p and two non-sister chromatids are chosen at random, and recombination occurs with probability r: the alleles at all loci distal to the crossing over position p are swapped between the two chromatids (therefore, I assume that crossing over always leads to recombination rather than just gene conversion). If r > 1 (in this case, r is an integer), a locus p and two non-sister chromatids are chosen at random, and recombination occurs with probability 1: the alleles at all loci distal to the crossing-over position p are swapped between the two chromatids; the process is repeated for a total of r times choosing a different locus (no chromosome interference) but with the same chromatids (chiasmata are reciprocal).
- Segregation of chromatids occurs: for mitosis, x segregation (in which each recombinant chromatid segregates with the non-sister non-recombinant chromatid) occurs with probability x, and z segregation (in which the two recombinant chromatids segregate together, apart from the two non-recombinant chromatids) occurs with probability 1-x.
- One random daughter cell produced by this process is chosen to replace the original cell.
- The process 1–7 is repeated for 1000 cells.
- The process 1–8 is repeated for 200 generations or until a stable value of LOH is reached (that is, the average over 50 generations does not change by more than 1%).
- The average value of LOH and fitness is measured as the average over the last 50 generations.
- The process 1–10 is repeated 10 times, and the results are averaged.
Funding
Conflicts of Interest
References
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [Green Version]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienta, K.J.; Hammarlund, E.U.; Austin, R.H.; Axelrod, R.; Brown, J.S.; Amend, S.R. Cancer cells employ an evolutionarily conserved polyploidization program to resist therapy. Semin. Cancer Biol. 2022, 81, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Haroske, G.; Baak, J.P.; Danielsen, H.; Giroud, F.; Gschwendtner, A.; Oberholzer, M.; Reith, A.; Spieler, P.; Böcking, A. Fourth updated ESACP consensus report on diagnostic DNA image cytometry. Anal. Cell Pathol. 2001, 23, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höglund, M.; Gisselsson, D.; Säll, T.; Mitelman, F. Coping with complexity. multivariate analysis of tumor karyotypes. Cancer Genet. Cytogenet. 2002, 135, 103–109. [Google Scholar] [CrossRef]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [Green Version]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Cancer: A matter of life cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Carballo, D.; Gustmann, S.; Jastrow, H.; Acikelli, A.H.; Dammann, P.; Klein, J.; Dembinski, U.; Bardenheuer, W.; Malak, S.; Araúzo-Bravo, M.J.; et al. Atypical cell populations associated with acquired resistance to cytostatics and cancer stem cell features: The role of mitochondria in nuclear encapsulation. DNA Cell Biol. 2014, 33, 749–774. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, D.; Mandal, M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020, 52, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [Green Version]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the aneuploidy and senescence paradoxes: A study of triploid breast cancers non-responsive to neoadjuvant therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, B.; Ghosh, D.; Dawson, M.R. Contributions of the distinct biophysical phenotype of polyploidal giant cancer cells to cancer progression. Semin. Cancer Biol. 2022, 81, 64–72. [Google Scholar] [CrossRef]
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.; Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Miroshnychenko, D.; Baratchart, E.; Ferrall-Fairbanks, M.C.; Velde, R.V.; Laurie, M.A.; Bui, M.M.; Tan, A.C.; Altrock, P.M.; Basanta, D.; Marusyk, A. Spontaneous cell fusions as a mechanism of parasexual recombination in tumour cell populations. Nat. Ecol. Evol. 2021, 5, 379–391. [Google Scholar] [CrossRef]
- Riffell, J.L.; Zimmerman, C.; Khong, A.; McHardy, L.M.; Roberge, M. Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle 2009, 8, 3025–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Kalejs, M.; Erenpreisa, J. Cancer/testis antigens and gametogenesis: A review and “brain-storming” session. Cancer Cell Int. 2005, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Kitano, H.; Takikita, M.; Cho, H.; Noh, K.H.; Kim, T.W.; Ylaya, K.; Hanaoka, J.; Fukuoka, J.; Hewitt, S.M. Synaptonemal complex protein 3 as a novel prognostic marker in early stage non-small cell lung cancer. Hum. Pathol. 2013, 44, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. J. Skin Cancer. 2013, 2013, 190109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Senovilla, L.; Jemaà, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Old, L.J. Cancer/testis (CT) antigens-a new link between gametogenesis and cancer. Cancer Immun. 2001, 1, 1. [Google Scholar] [PubMed]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Liloglou, T.; Kokotas, S.; Kastrinakis, N.; Kotsinas, A.; Athanasiou, A.; Foukas, P.; Zoumpourlis, V.; et al. Deregulated expression of c-mos in non-small cell lung carcinomas: Relationship with p53 status, genomic instability, and tumor kinetics. Cancer Res. 2001, 61, 538–549. [Google Scholar]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Santoro, I.M.; McDaniel, L.D.; Nishijima, I.; Mills, M.; Youssoufian, H.; Vogel, H.; Schultz, R.A.; Bradley, A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 2000, 26, 424–429. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, R.J.; Wakeman, J.A. Meiosis-like Functions in Oncogenesis: A New View of Cancer. Cancer Res. 2017, 77, 5712–5716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2014, 2, 3–14. [Google Scholar] [CrossRef]
- Archetti, M. Recombination and loss of complementation: A more than two-fold cost for parthenogenesis. J. Evol. Biol. 2004, 17, 1084–1097. [Google Scholar] [CrossRef]
- Archetti, M. Loss of complementation and the logic of two-step meiosis. J. Evol. Biol. 2004, 17, 1098–1105. [Google Scholar] [CrossRef]
- Archetti, M. Complementation, genetic conflict, and the evolution of sex and recombination. J. Hered. 2010, 101 (Suppl. S1), S21–S33. [Google Scholar] [CrossRef] [Green Version]
- Archetti, M. Inverted meiosis and the evolution of sex by loss of complementation. J. Evol. Biol. 2020, 33, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Evidence from automixis with inverted meiosis for the maintenance of sex by loss of complementation. J. Evol. Biol. 2022, 35, 40–50. [Google Scholar] [CrossRef]
- Stern, C. Somatic Crossing over and Segregation in Drosophila Melanogaster. Genetics 1936, 21, 625–730. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Antioncogenes and human cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 10914–10921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavenee, W.K.; Dryja, T.P.; Phillips, R.A.; Benedict, W.F.; Godbout, R.; Gallie, B.L.; Murphree, A.L.; Strong, L.C.; White, R.L. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983, 305, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M.; Ferraro, D.A.; Christofori, G. Heterogeneity for IGF-II production maintained by public goods dynamics in neuroendocrine pancreatic cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, L.G.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.L.; Leith, J.T. Tumour heterogeneity and drug resistance. J. Clin. Oncol. 1986, 4, 244–257. [Google Scholar] [CrossRef]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.-A.; Rabinovitch, P.S.; et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar] [CrossRef]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Annu. Rev. Pathol. Mech. 2013, 8, 277–302. [Google Scholar] [CrossRef] [Green Version]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; WM Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Vosoughi, A.; Zhang, T.; Shohdy, K.S.; Vlachostergios, P.J.; Wilkes, D.C.; Bhinder, B.; Tagawa, S.T.; Nanus, D.M.; Molina, A.M.; Beltran, H.; et al. Common germline-somatic variant interactions in advanced urothelial cancer. Nat. Commun. 2020, 11, 6195. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic. Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Systemic evolutionary changes in mammalian gene expression. Biosystems 2020, 198, 104256. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef]
- Tischfield, J.A. Loss of heterozygosity or: How I learned to stop worrying and love mitotic recombination. Am. J. Hum. Genet. 1997, 61, 995–999. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef]
- Zhang, X.; Sjöblom, T. Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals 2021, 14, 57. [Google Scholar] [CrossRef]
- Rendo, V.; Stoimenov, I.; Mateus, A.; Sjöberg, E.; Svensson, R.; Gustavsson, A.L.; Johansson, L.; Ng, A.; OʼBrien, C.; Giannakis, M.; et al. Exploiting loss of heterozygosity for allele-selective colorectal cancer chemotherapy. Nat. Commun. 2020, 11, 1308. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.S.; Mog, B.J.; Douglass, J.; Pearlman, A.H.; Hsiue, E.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Pardoll, D.M.; Gabelli, S.B.; et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc. Natl. Acad. Sci. USA 2021, 118, e2022410118. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Archetti, M. Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer. Int. J. Mol. Sci. 2022, 23, 8528. https://doi.org/10.3390/ijms23158528
Archetti M. Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer. International Journal of Molecular Sciences. 2022; 23(15):8528. https://doi.org/10.3390/ijms23158528
Chicago/Turabian StyleArchetti, Marco. 2022. "Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer" International Journal of Molecular Sciences 23, no. 15: 8528. https://doi.org/10.3390/ijms23158528
APA StyleArchetti, M. (2022). Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer. International Journal of Molecular Sciences, 23(15), 8528. https://doi.org/10.3390/ijms23158528