
Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
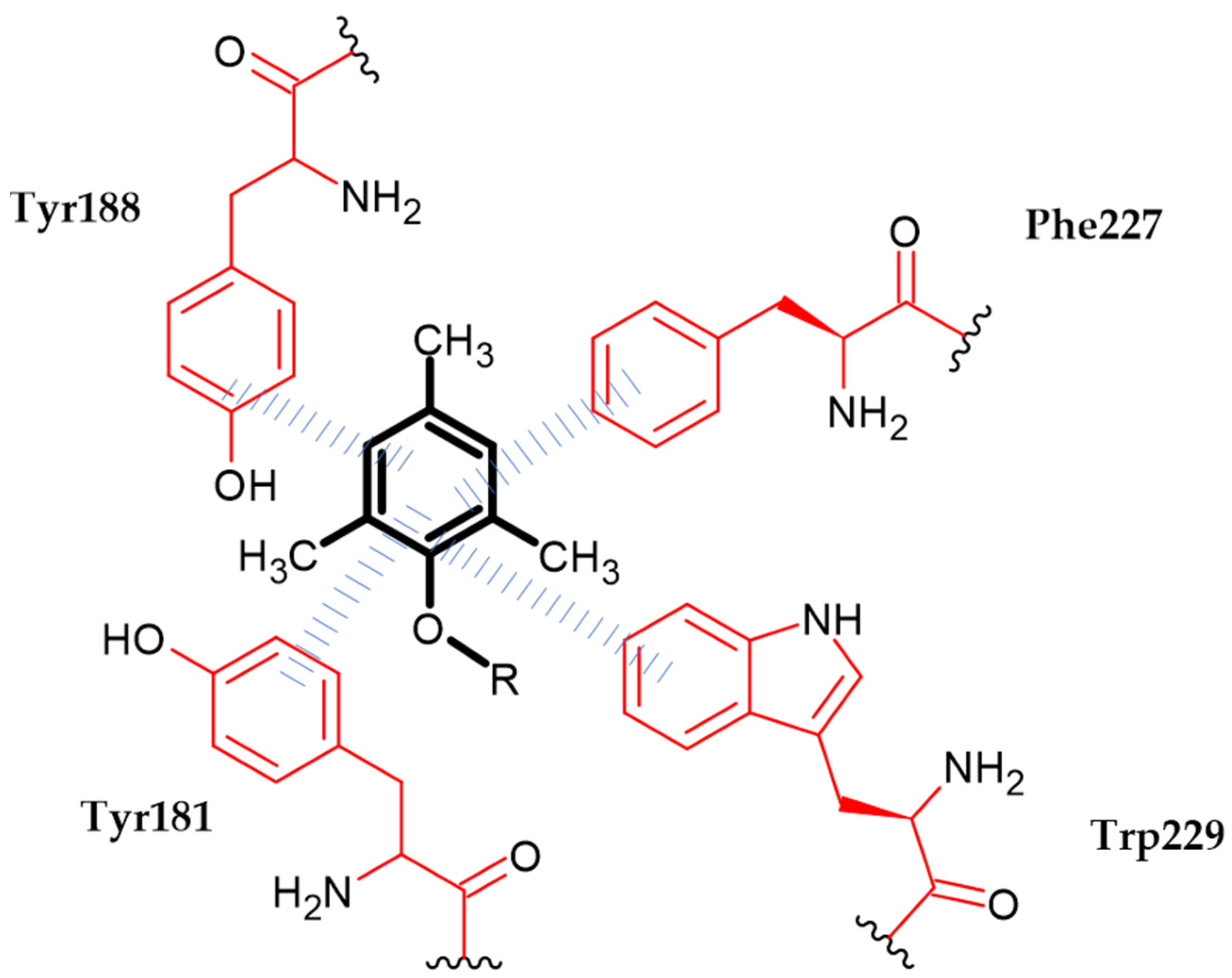{kind=link}
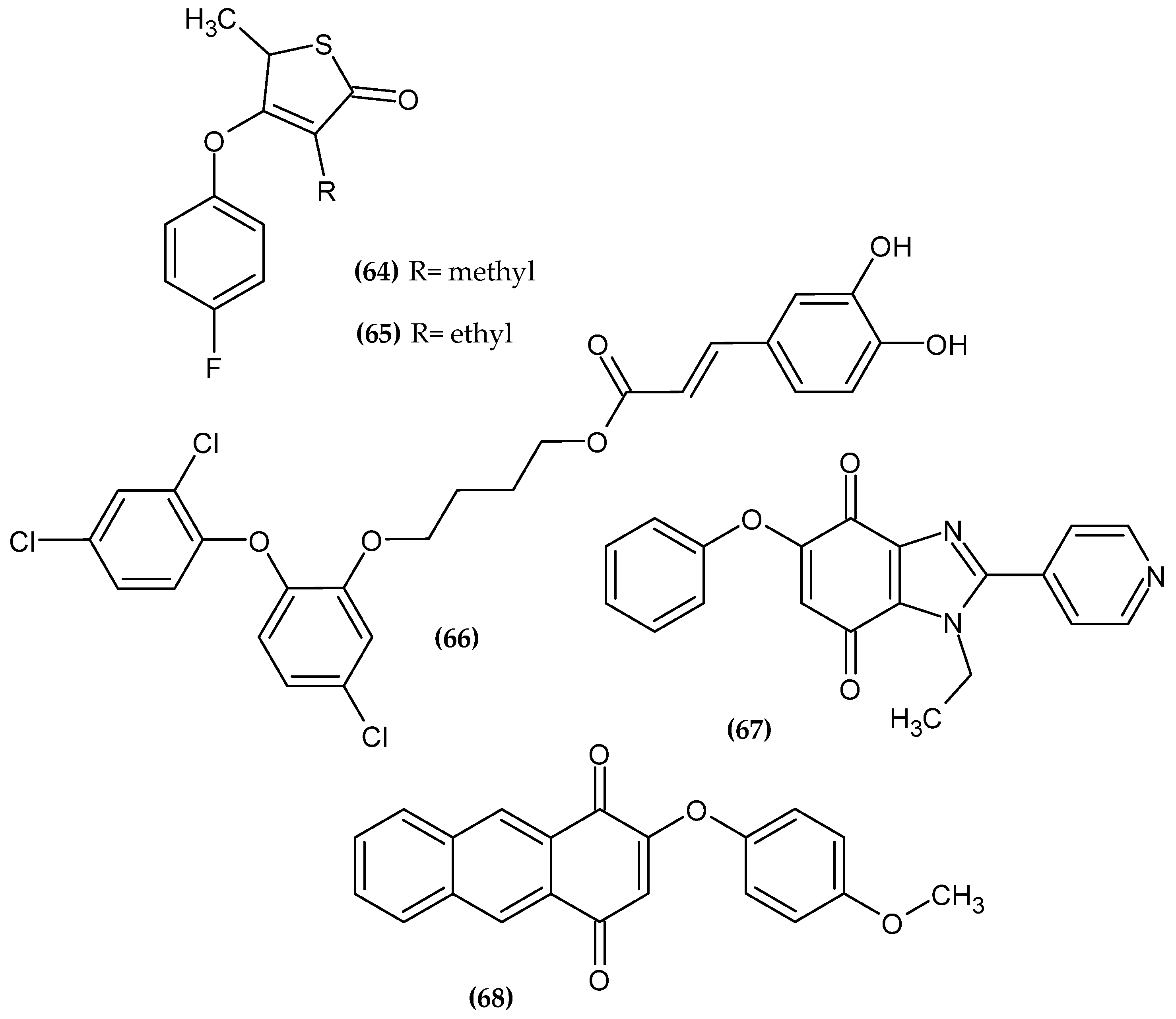{kind=link}
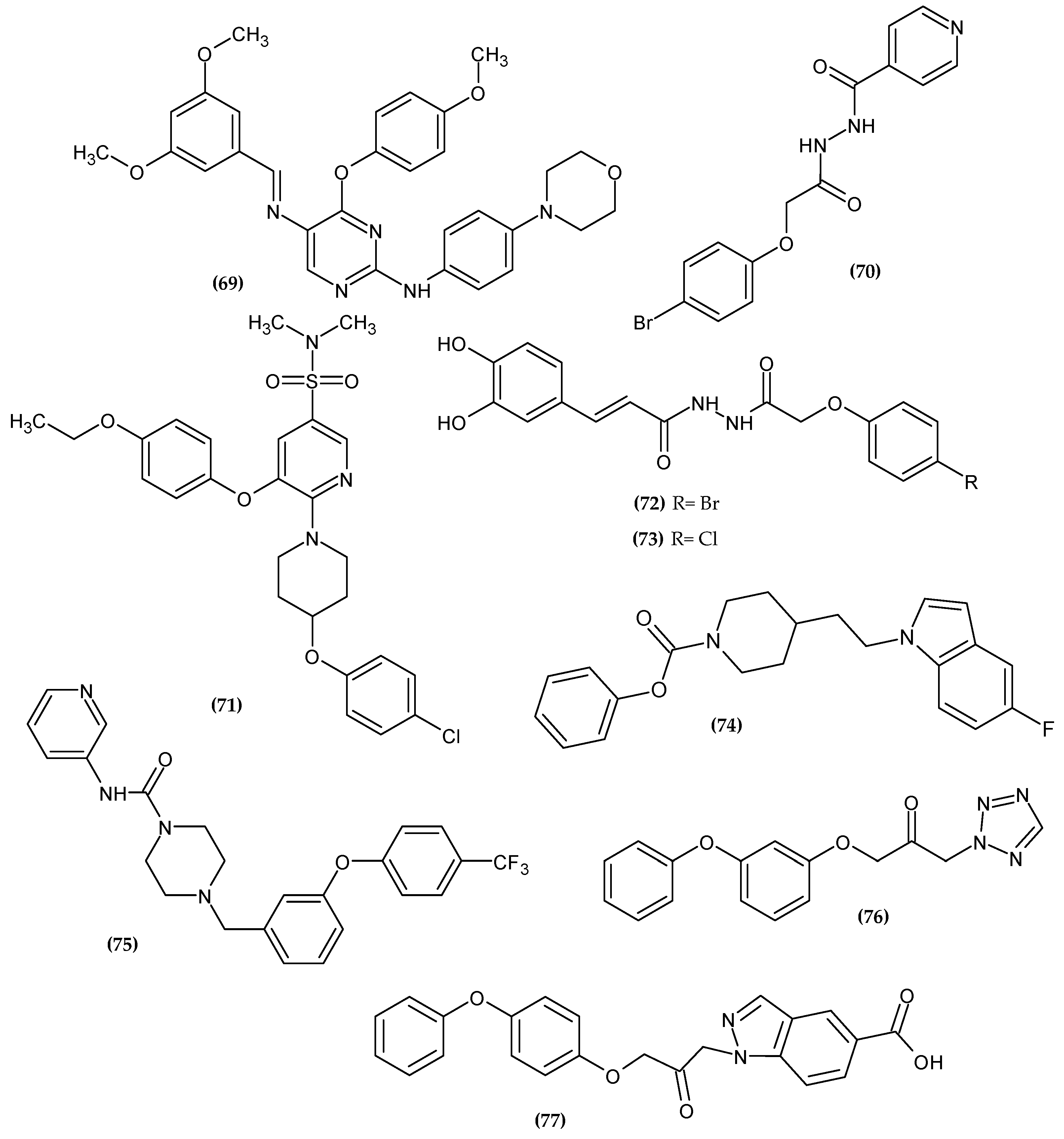{kind=link}
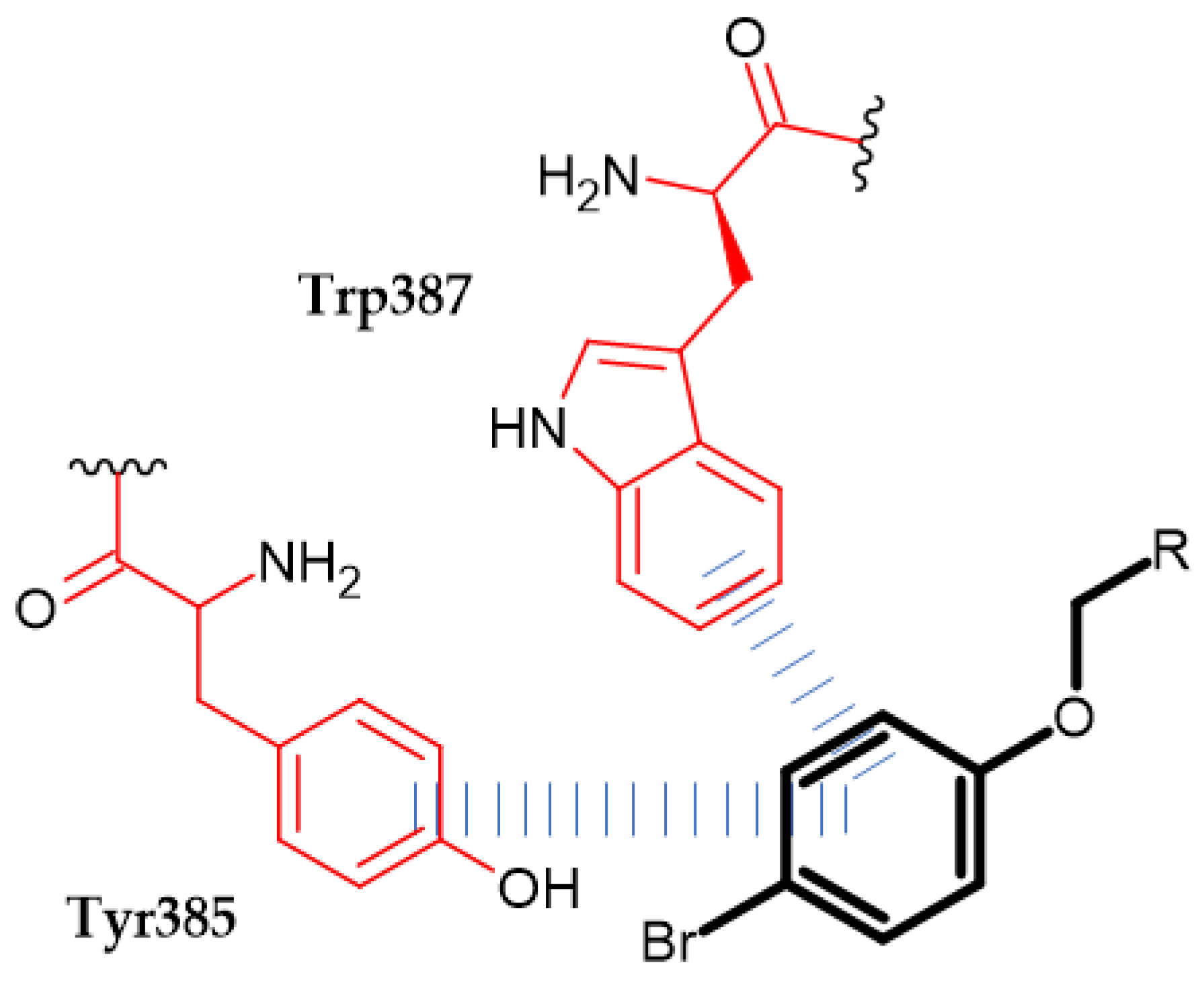{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
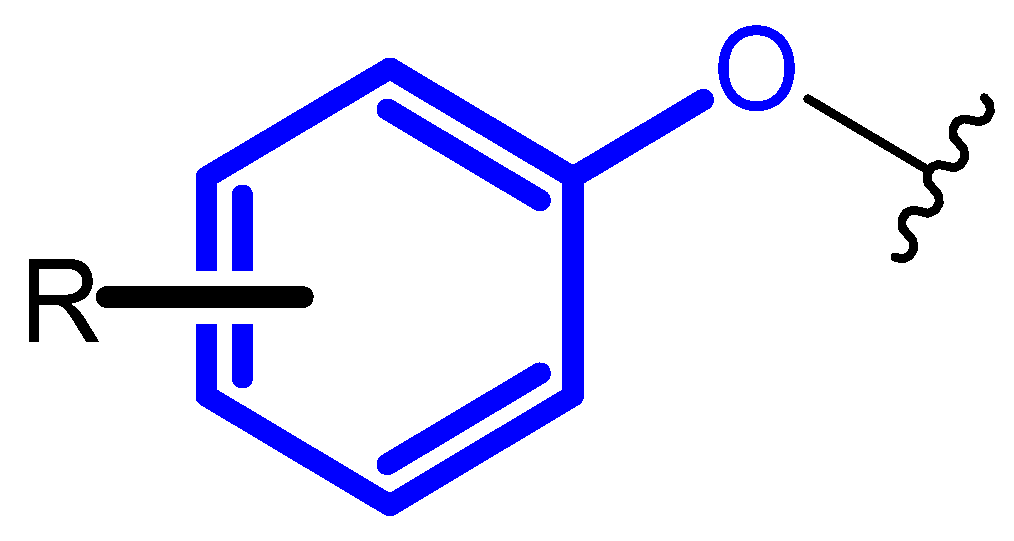
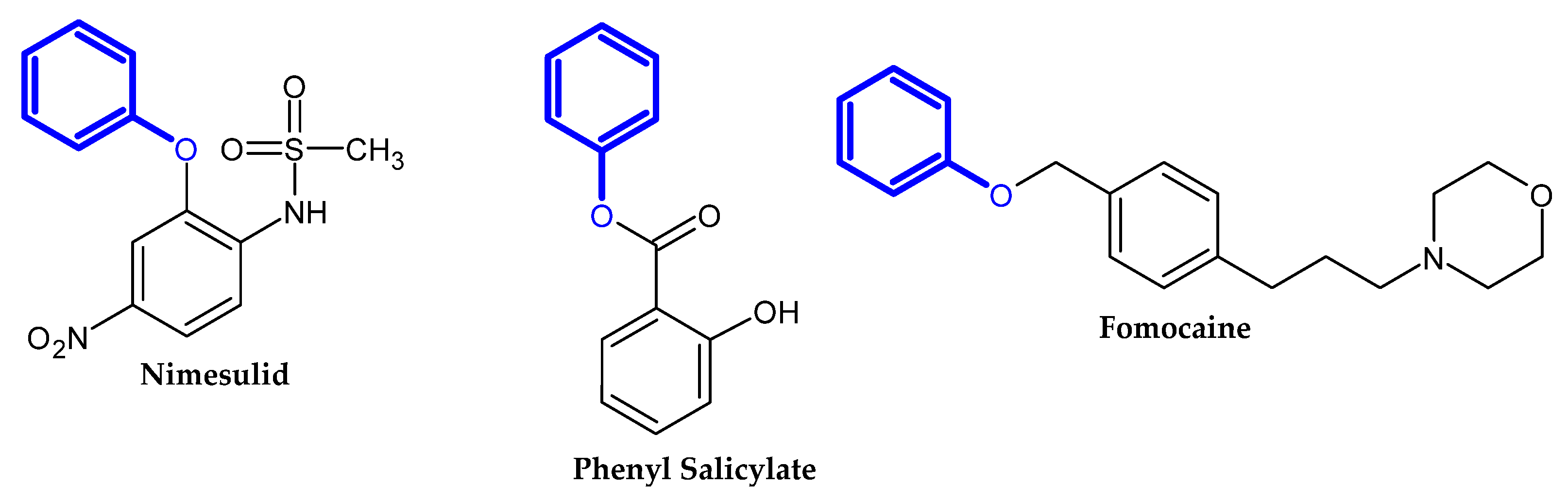
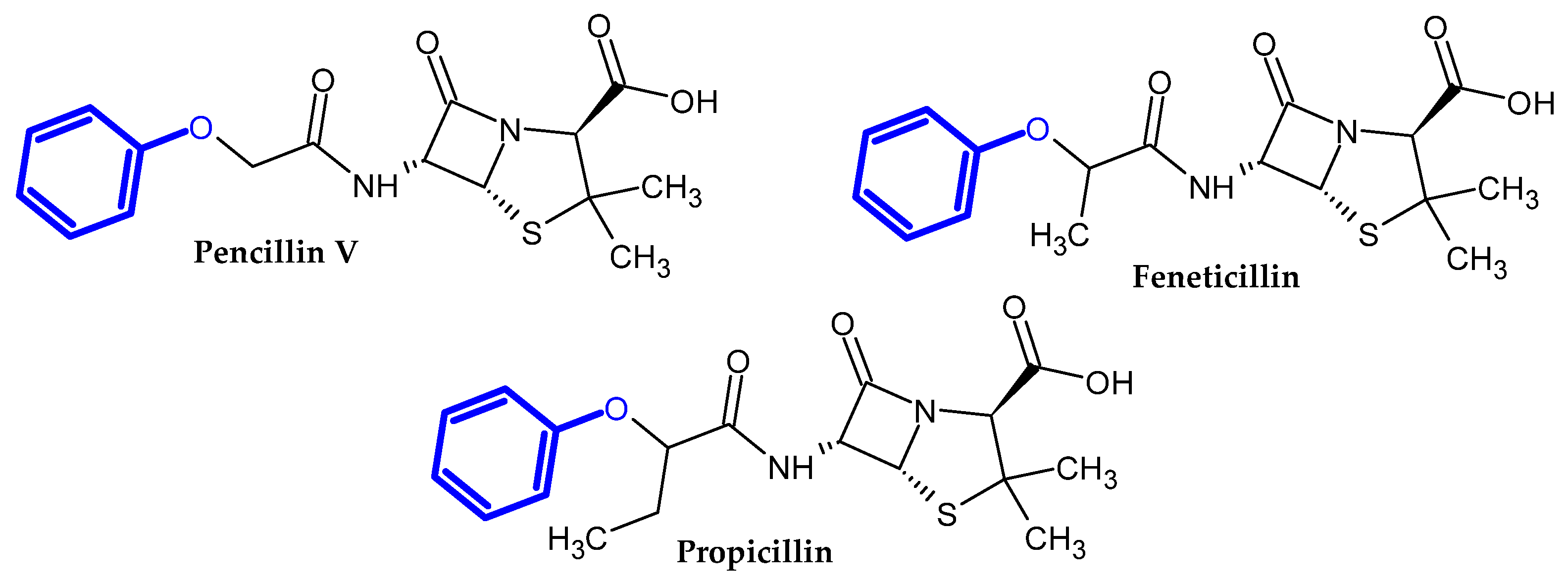
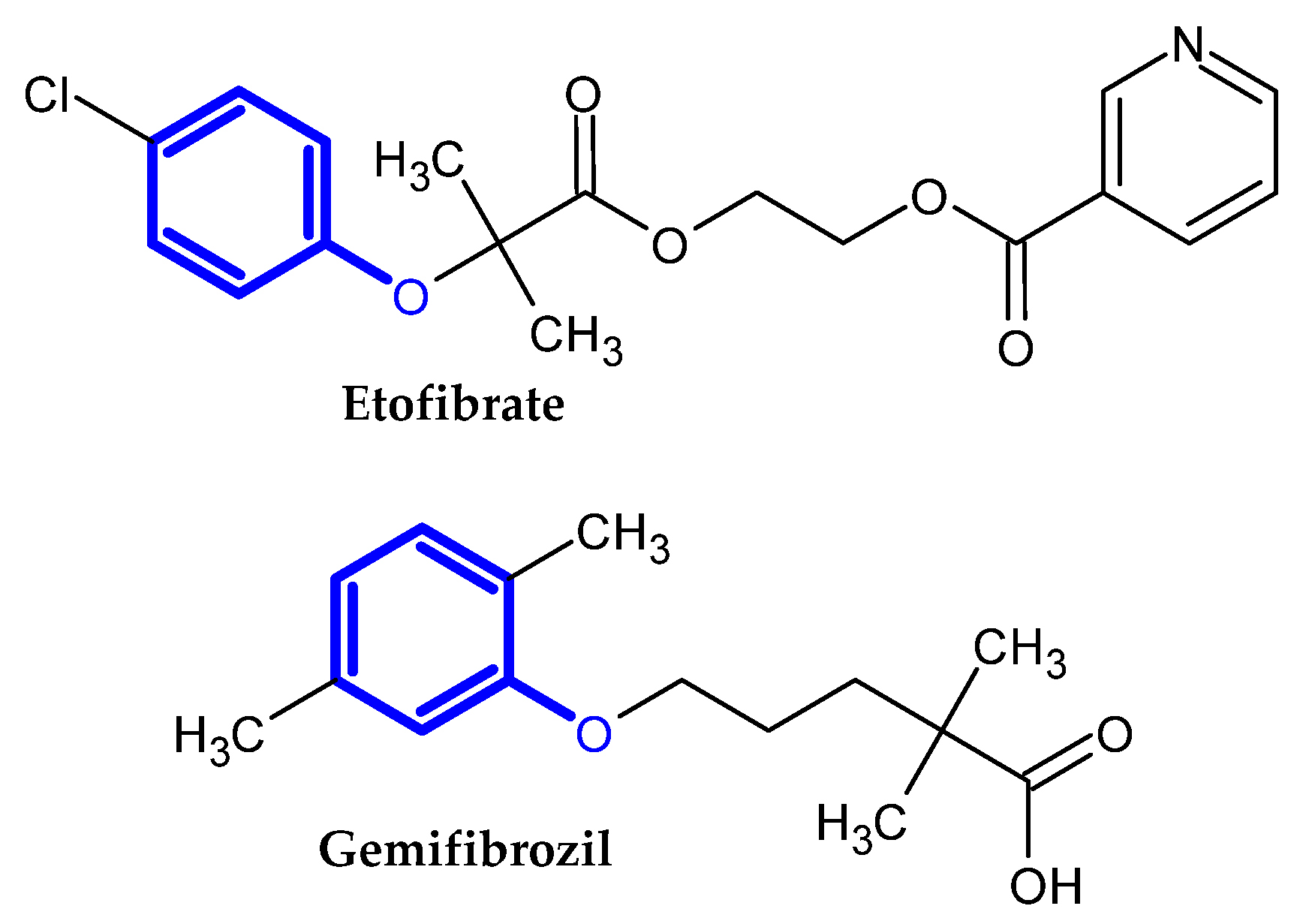
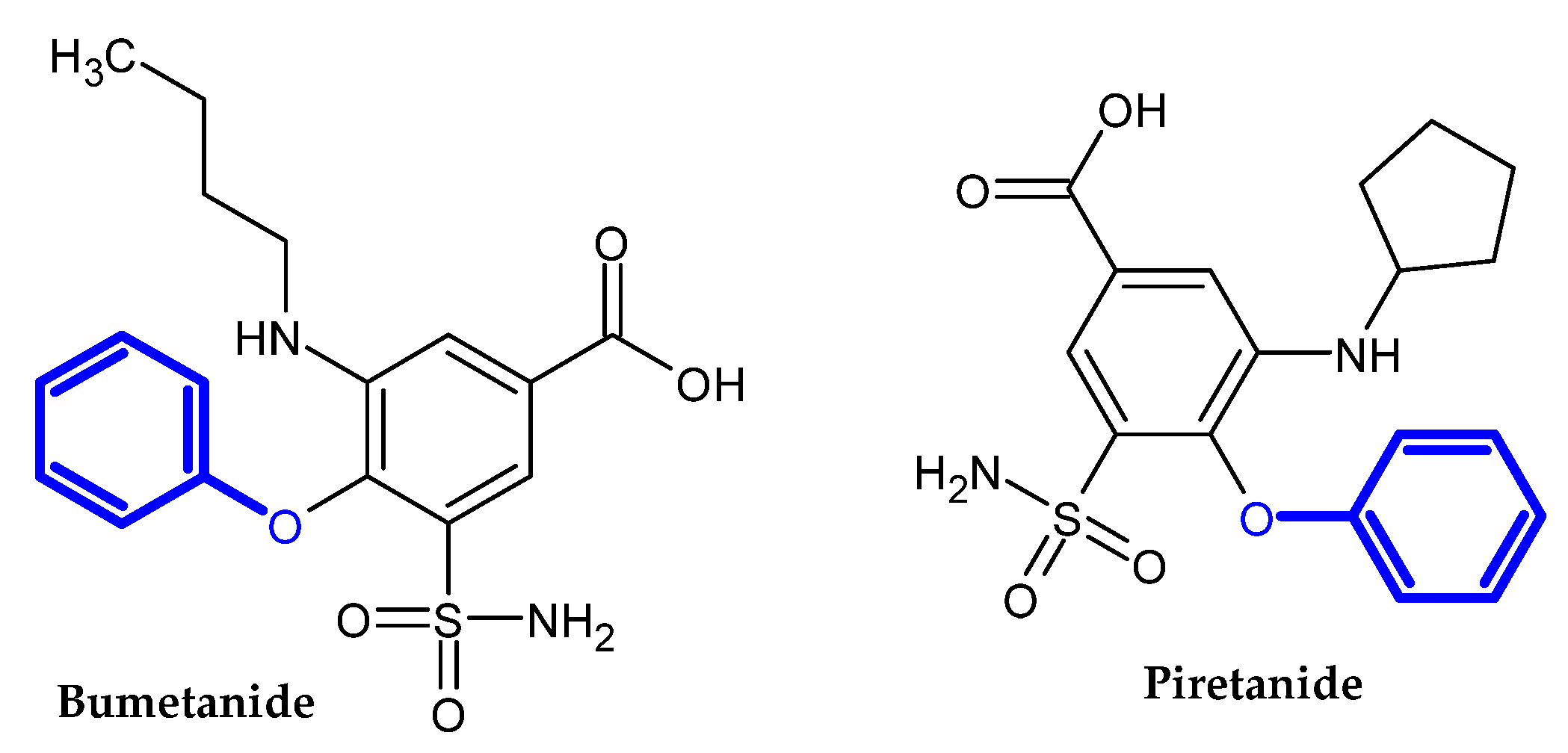
2. Compound Bearing Terminal Phenoxy Group Currently in Use
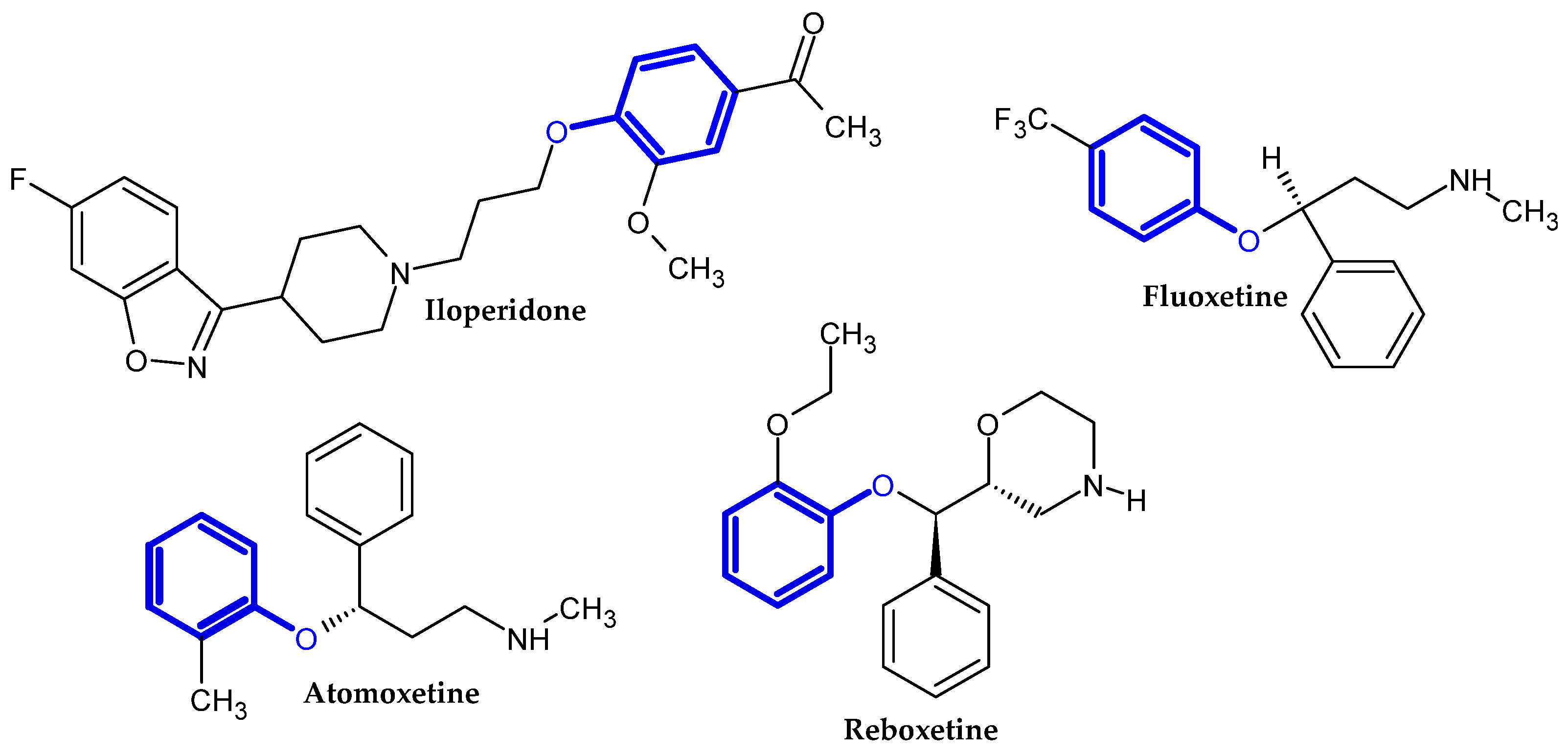
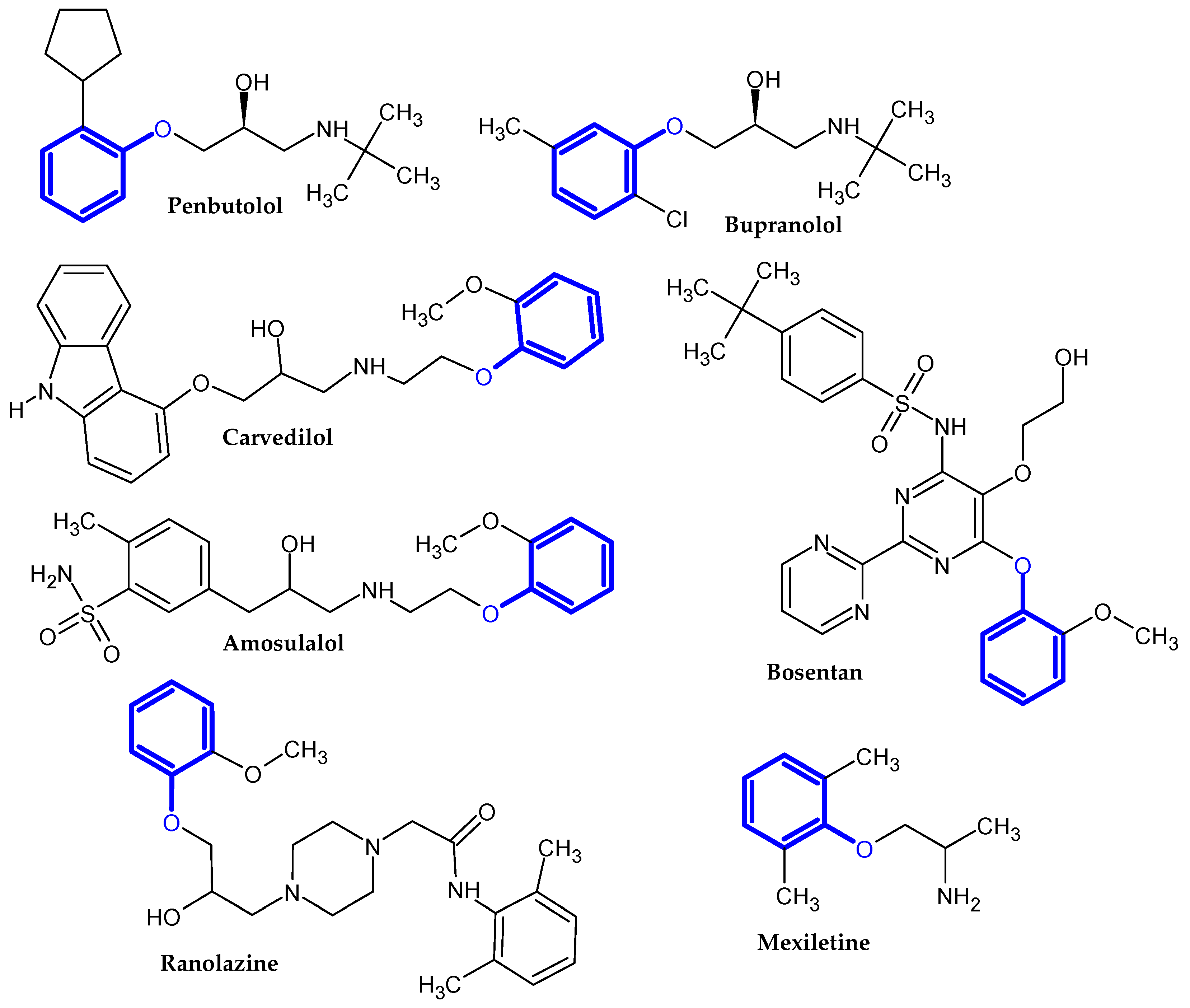
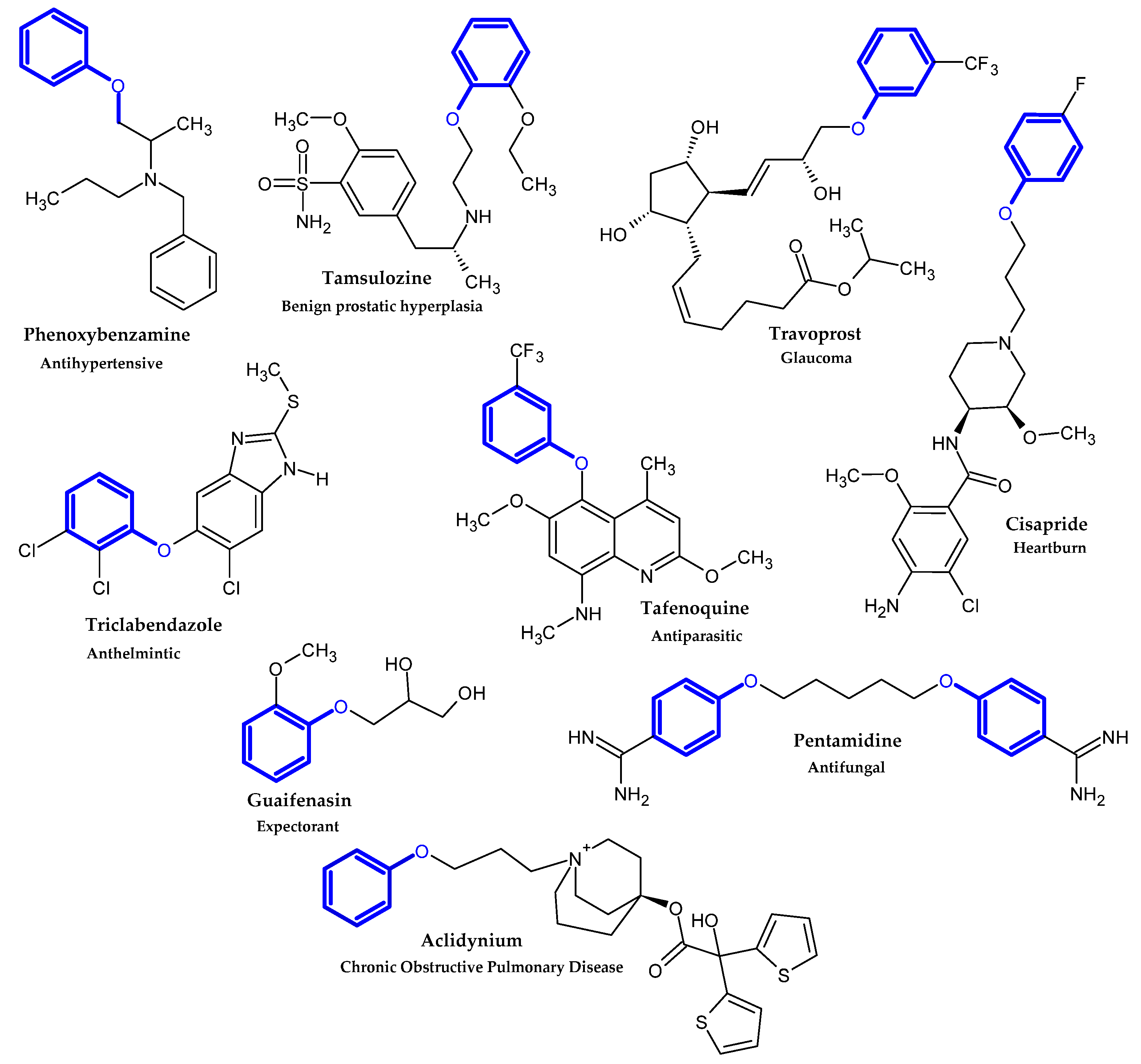
2.1. FDA-Approved Drugs
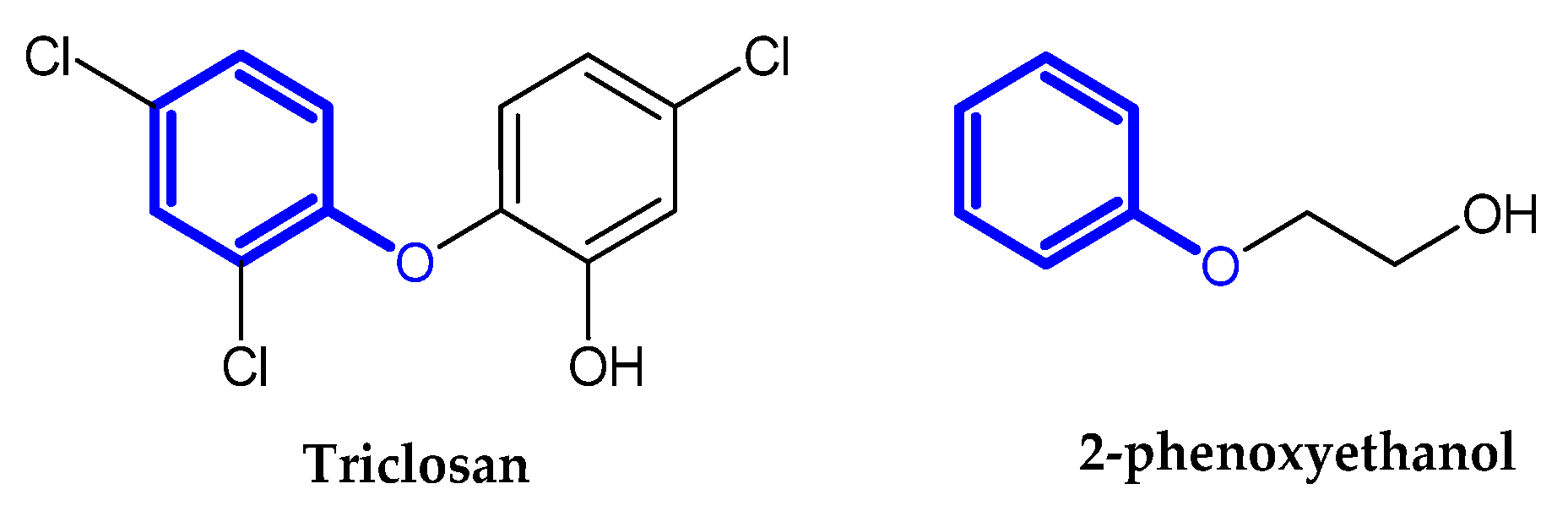
2.2. Auxiliary Substances Bearing the Phenoxy Group
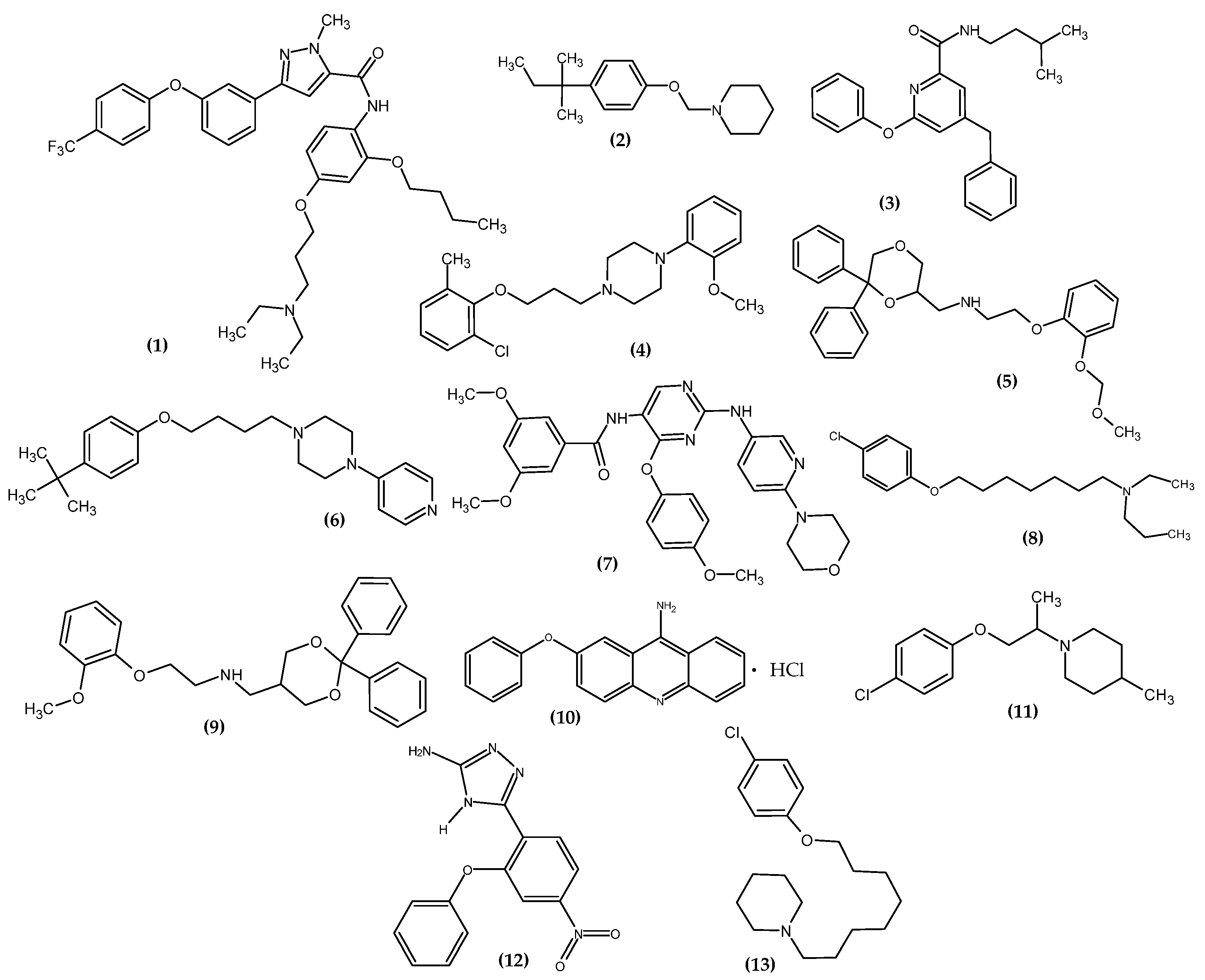
3. Novel Agent with the Terminal Phenoxy Group from the Most Recent Studies
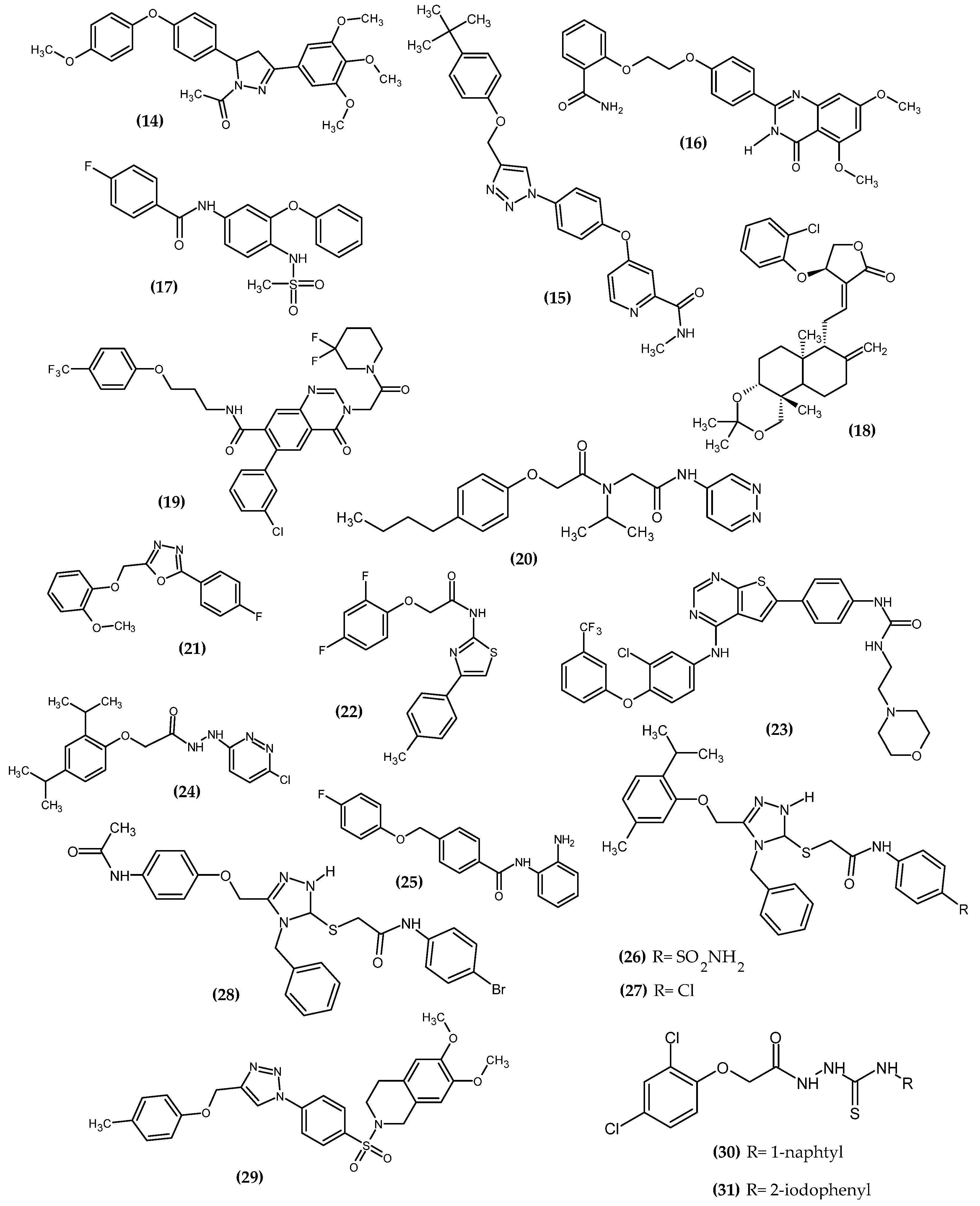
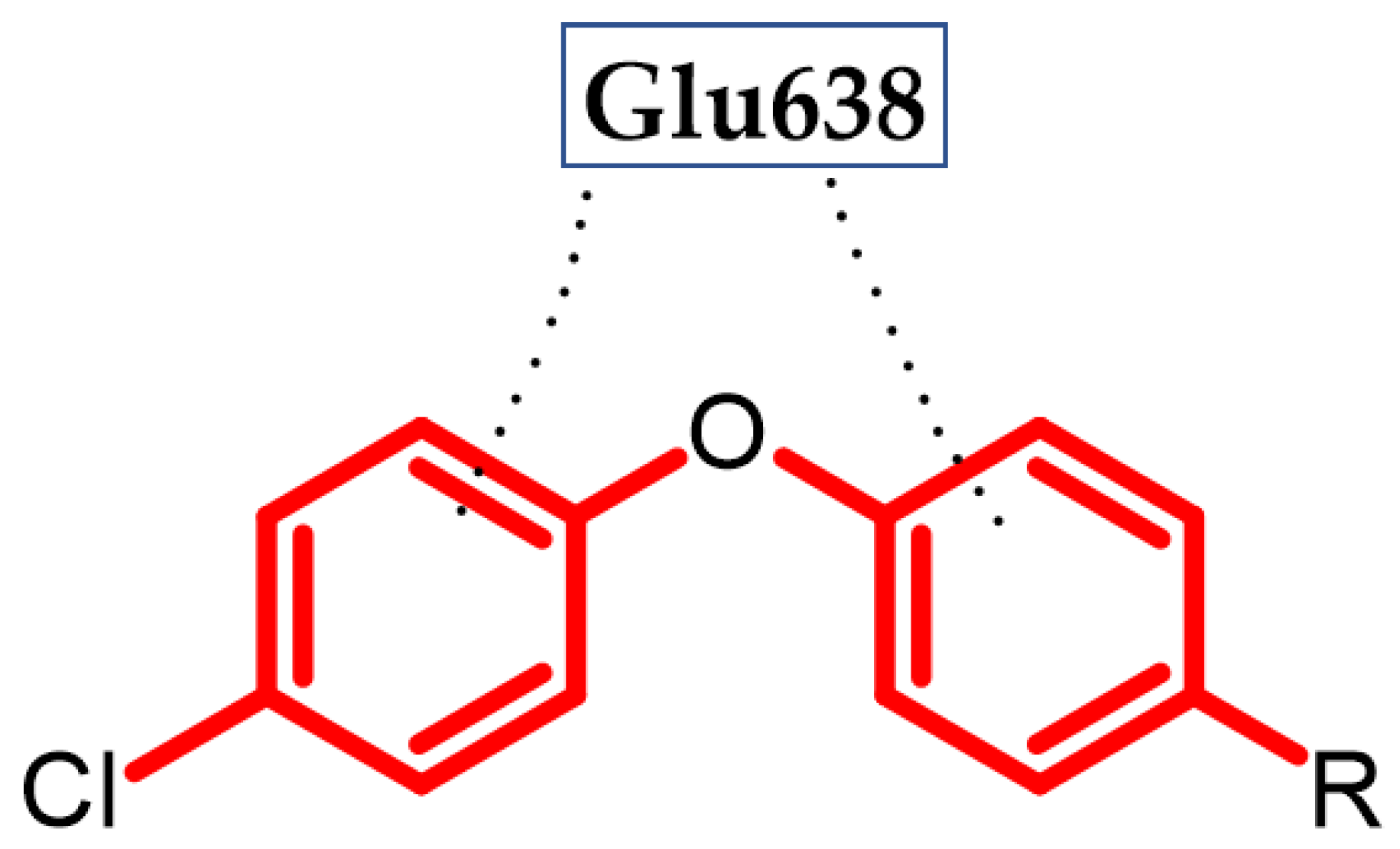
3.1. Neurological Disorders
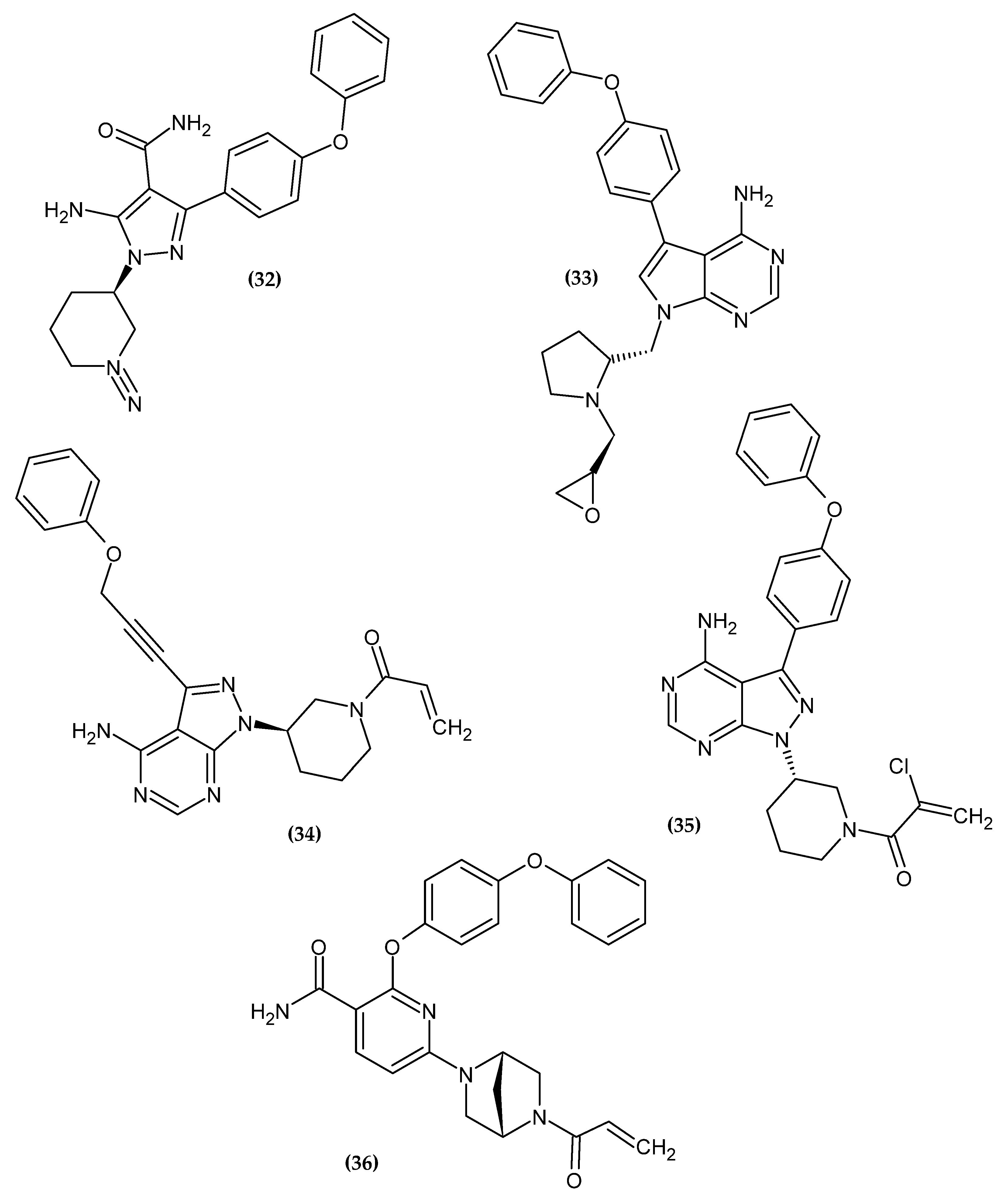
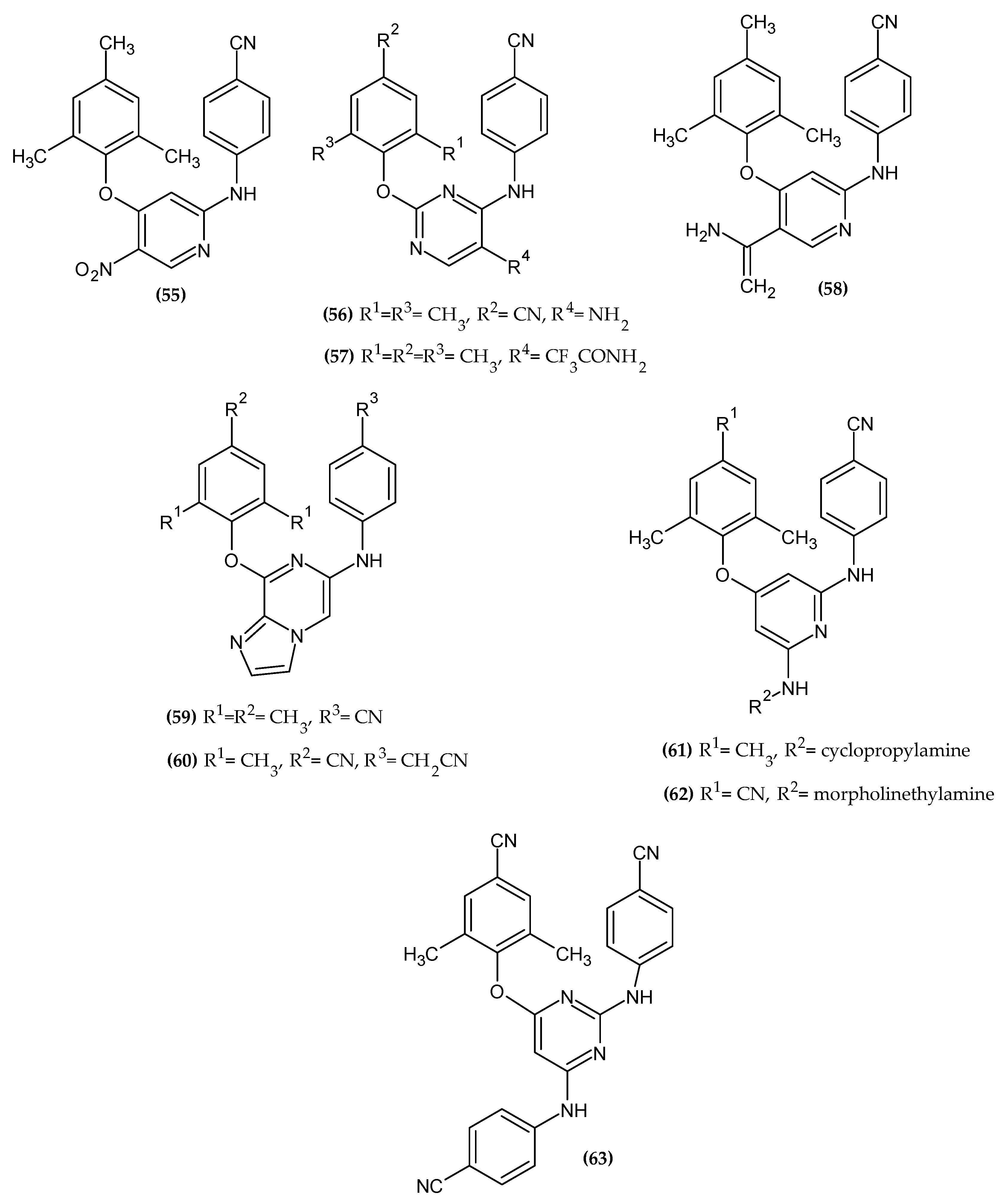
3.2. Anticancer Activity
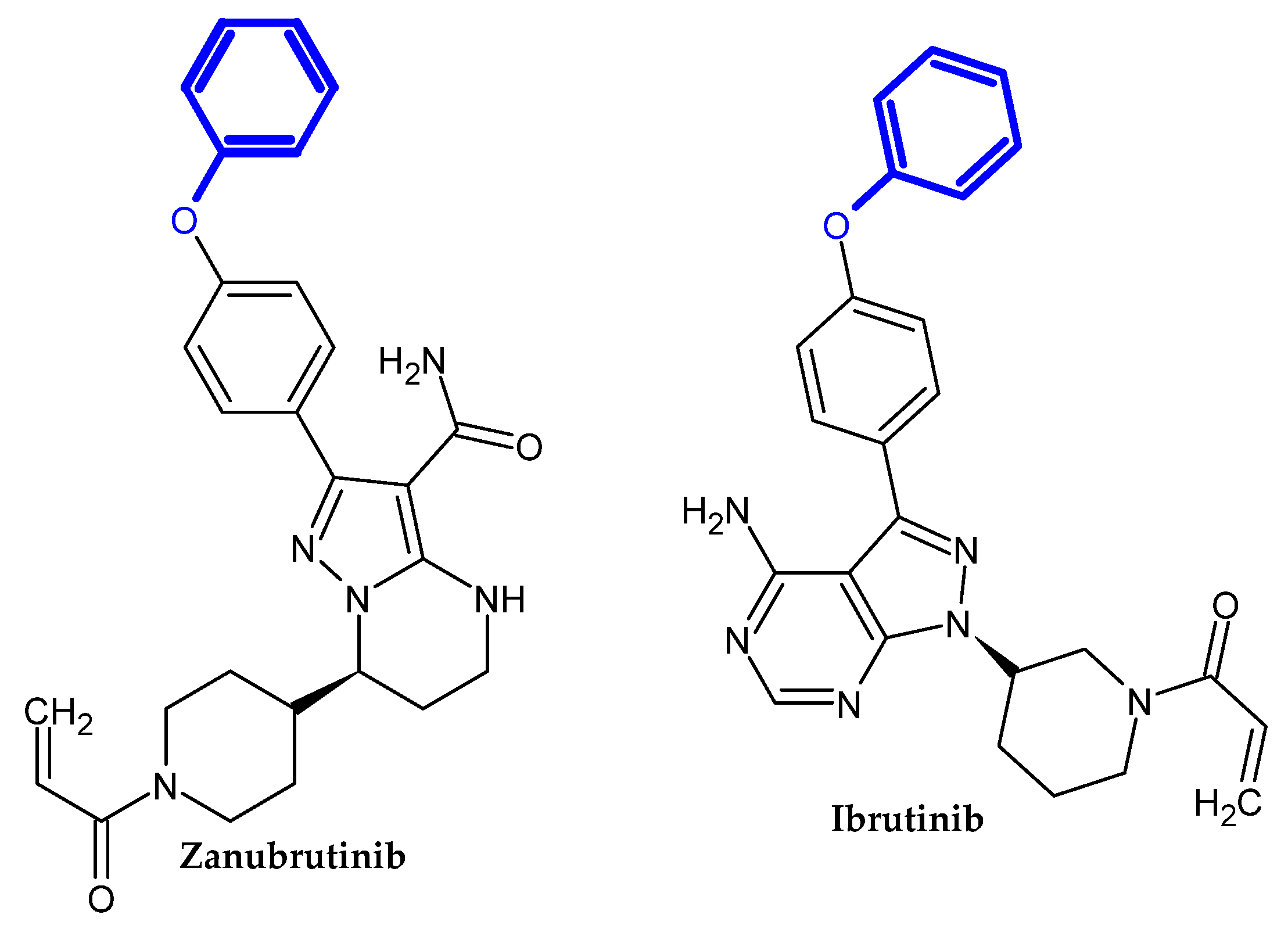
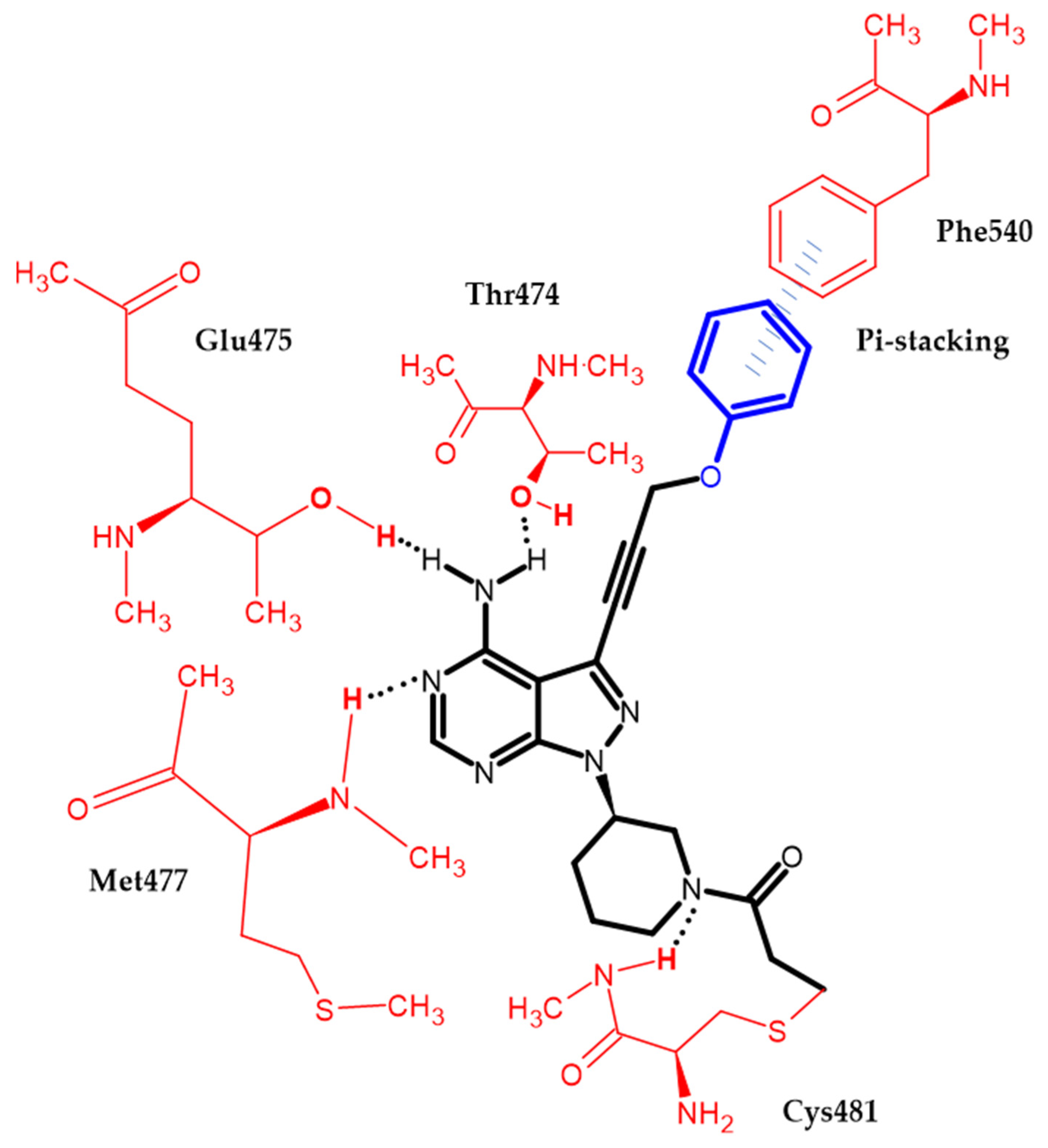
Bruton Tyrosine Kinase Inhibitors
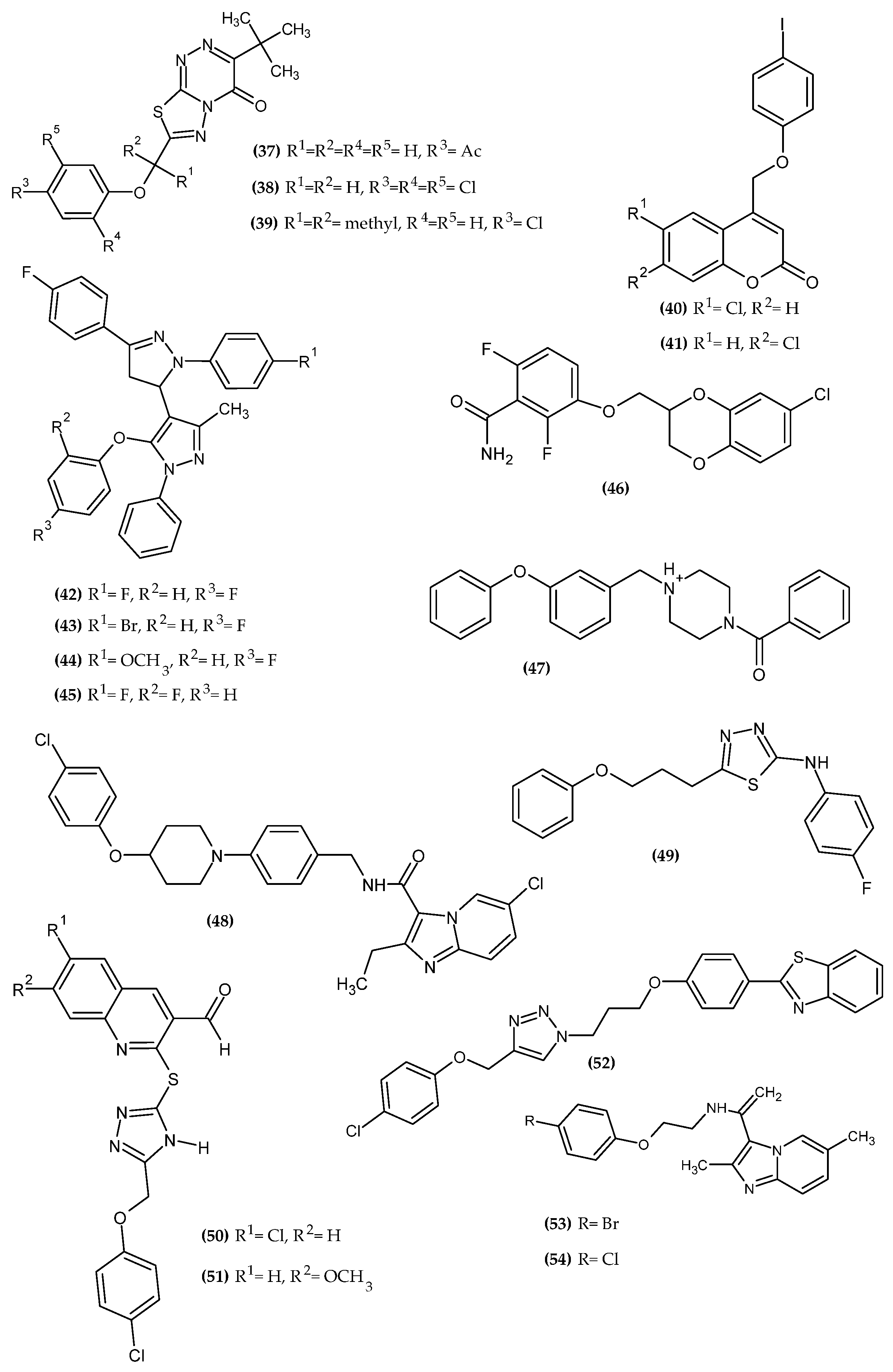
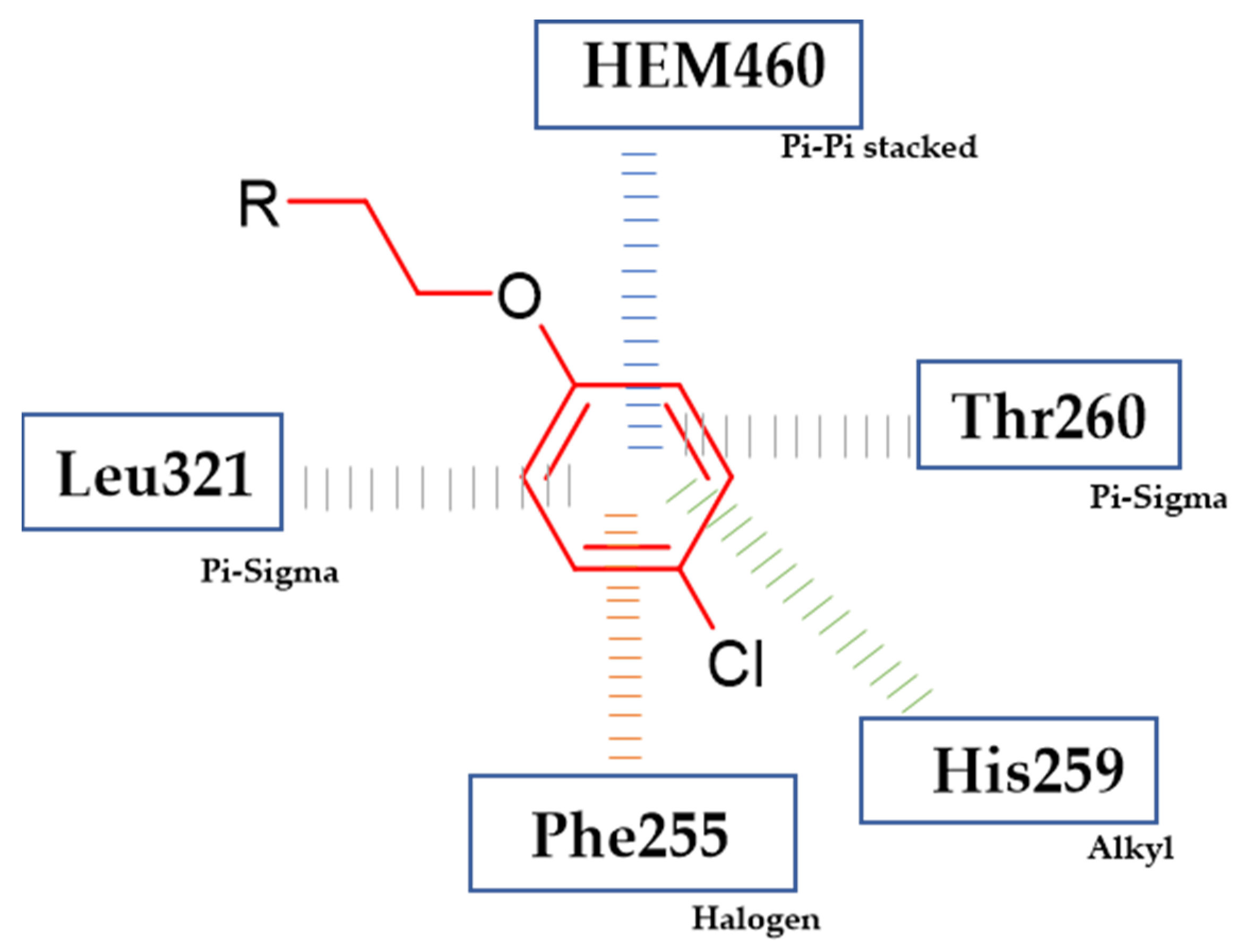
3.3. Antimicrobial Activity
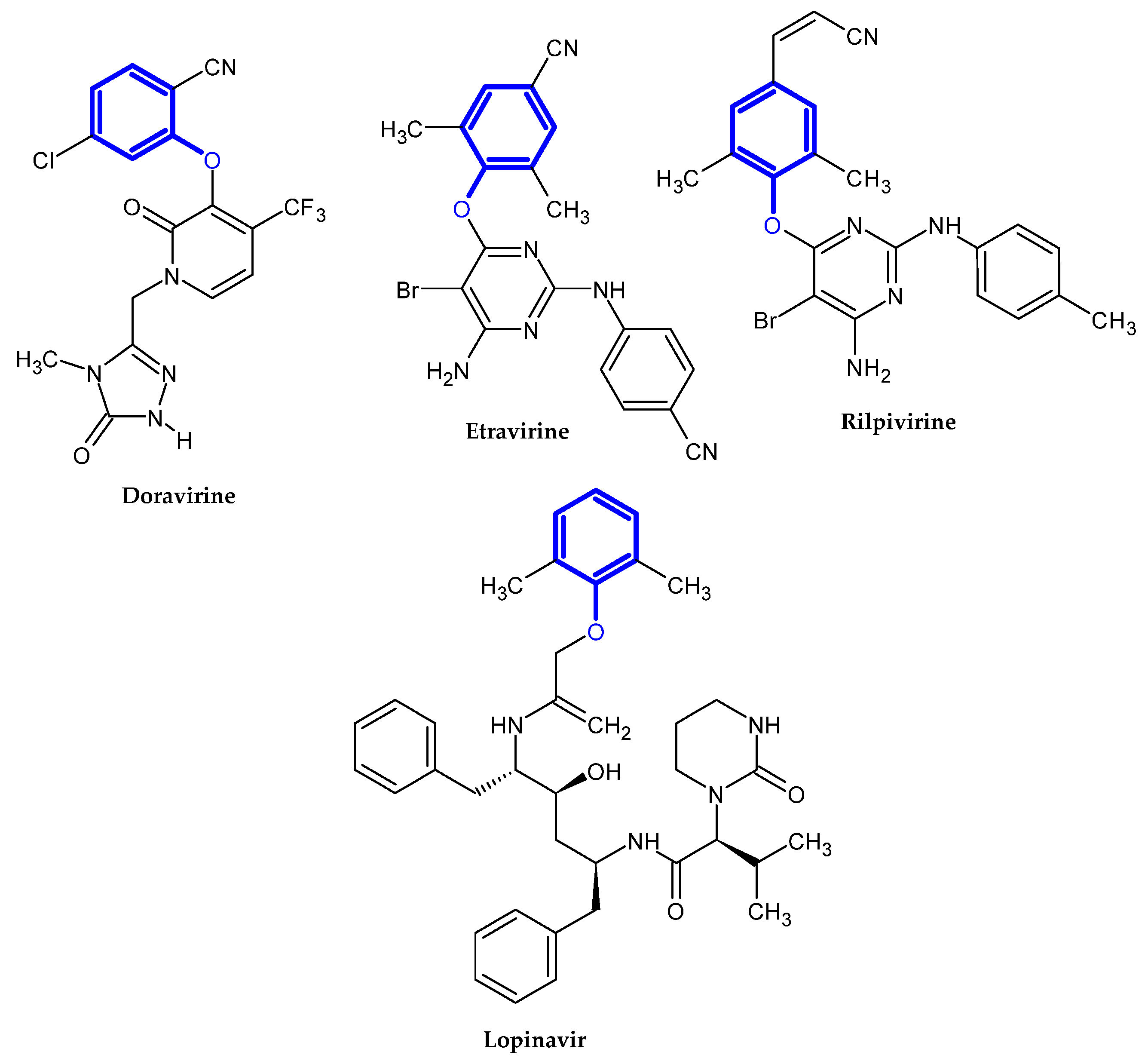
3.4. Anti-HIV Activity
3.5. Antiparasitic Activity
3.6. Analgesic Activity
3.7. Anti-Diabetic Activity
3.8. Other Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mullard, A. 2021 FDA Approvals. Nat. Rev. Drug Discov. 2022, 21, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Phase II Trials in Drug Development and Adaptive Trial Design. JACC Basic Transl. Sci. 2019, 4, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.K. Phase II and Phase III Failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Spellberg, B.; Gilbert, D.N. The Future of Antibiotics and Resistance: A Tribute to a Career of Leadership by John Bartlett. Clin. Infect. Dis. 2014, 59, S71–S75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COVID Live—Coronavirus Statistics—Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 14 May 2022).
- Patrick, G.L. An Introduction to Medicinal Chemistry; Oxford University Press: Oxford, UK, 2017; ISBN 978-0-19-874969-1. [Google Scholar]
- Mao, F.; Ni, W.; Xu, X.; Wang, H.; Wang, J.; Ji, M.; Li, J. Chemical Structure-Related Drug-Like Criteria of Global Approved Drugs. Molecules 2016, 21, 75. [Google Scholar] [CrossRef] [Green Version]
- Deb, P.K.; Al-Attraqchi, O.; Jaber, A.Y.; Amarji, B.; Tekade, R.K. Chapter 2—Physicochemical Aspects to Be Considered in Pharmaceutical Product Development. In Dosage Form Design Considerations; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2018; pp. 57–83. ISBN 978-0-12-814423-7. [Google Scholar]
- Yang, J.; Chen, W.; Kang, D.; Lu, X.; Li, X.; Liu, Z.; Huang, B.; Daelemans, D.; Pannecouque, C.; De Clercq, E.; et al. Design, Synthesis and Anti-HIV Evaluation of Novel Diarylpyridine Derivatives Targeting the Entrance Channel of NNRTI Binding Pocket. Eur. J. Med. Chem. 2016, 109, 294–304. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, W.; Zhan, P.; De Clercq, E.; Pannecouque, C.; Liu, X. Design, Synthesis and Anti-HIV Evaluation of Novel Diarylnicotinamide Derivatives (DANAs) Targeting the Entrance Channel of the NNRTI Binding Pocket through Structure-Guided Molecular Hybridization. Eur. J. Med. Chem. 2014, 87, 52–62. [Google Scholar] [CrossRef]
- Lowe, F.C. Summary of Clinical Experiences with Tamsulosin for the Treatment of Benign Prostatic Hyperplasia. Rev. Urol. 2005, 7 (Suppl. 4), S13–S21. [Google Scholar]
- Liu, J.; Chen, C.; Wang, D.; Zhang, J.; Zhang, T. Emerging Small-Molecule Inhibitors of the Bruton’s Tyrosine Kinase (BTK): Current Development. Eur. J. Med. Chem. 2021, 217, 113329. [Google Scholar] [CrossRef]
- Khanum, S.A.; Khanum, N.F.; Shashikanth, M. Synthesis and Anti-Inflammatory Activity of 2-Aryloxy Methyl Oxazolines. Bioorg. Med. Chem. Lett. 2008, 18, 4597–4601. [Google Scholar] [CrossRef] [PubMed]
- Dréno, B.; Zuberbier, T.; Gelmetti, C.; Gontijo, G.; Marinovich, M. Safety Review of Phenoxyethanol When Used as a Preservative in Cosmetics. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Heath, R.J.; Rubin, J.R.; Holland, D.R.; Zhang, E.; Snow, M.E.; Rock, C.O. Mechanism of Triclosan Inhibition of Bacterial Fatty Acid Synthesis. J. Biol. Chem. 1999, 274, 11110–11114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Hou, N.-N.; Wu, H.-M.; Zuo, X.; Lian, Y.-Z.; Zhang, C.-N.; Wang, Z.-F.; Zhang, X.; Zhu, J.-H. Prevalence of Alzheimer’s Disease and Parkinson’s Disease in China: An Updated Systematical Analysis. Front. Aging Neurosci. 2020, 12, 603854. [Google Scholar] [CrossRef] [PubMed]
- White, J. Please Remember the Real Me When I Cannot Remember You. Neurodegener. Dis. Manag. 2020, 10, 339–341. [Google Scholar] [CrossRef]
- Han, Y.T.; Kim, K.; Choi, G.-I.; An, H.; Son, D.; Kim, H.; Ha, H.-J.; Son, J.-H.; Chung, S.-J.; Park, H.-J.; et al. Pyrazole-5-Carboxamides, Novel Inhibitors of Receptor for Advanced Glycation End Products (RAGE). Eur. J. Med. Chem. 2014, 79, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Kuder, K.J.; Łażewska, D.; Kaleta, M.; Latacz, G.; Kottke, T.; Olejarz, A.; Karcz, T.; Fruziński, A.; Szczepańska, K.; Karolak-Wojciechowska, J.; et al. Synthesis and Biological Activity of Novel Tert-Amylphenoxyalkyl (Homo)Piperidine Derivatives as Histamine H3R Ligands. Bioorg. Med. Chem. 2017, 25, 2701–2712. [Google Scholar] [CrossRef]
- Tiligada, E.; Kyriakidis, K.; Chazot, P.L.; Passani, M.B. Histamine Pharmacology and New CNS Drug Targets. CNS Neurosci. Ther. 2011, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Stark, H. Histamine Receptor Subtypes: A Century of Rational Drug Design. Front. Biosci. 2012, 4, 461–488. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Modulation of In Vitro Neurotransmission in the CNS and in the Retina via H3 Heteroreceptors. In Pharmacochemistry Library; Leurs, R., Timmerman, H., Eds.; The Histamine H Receptor; Elsevier: Amsterdam, The Netherlands, 1998; Volume 30, pp. 13–26. [Google Scholar]
- Blandina, P.; Bacciottini, L.; Giovannini, M.G.; Mannaioni, P.F. H3 Receptor Modulation of the Release of Neurotransmitters In Vivo. In Pharmacochemistry Library; Leurs, R., Timmerman, H., Eds.; The Histamine H Receptor; Elsevier: Amsterdam, The Netherlands, 1998; Volume 30, pp. 27–40. [Google Scholar]
- Tiligada, E.; Zampeli, E.; Sander, K.; Stark, H. Histamine H3 and H4 Receptors as Novel Drug Targets. Expert Opin. Investig. Drugs 2009, 18, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Berlin, M.; Boyce, C.W.; de Lera Ruiz, M. Histamine H3 Receptor as a Drug Discovery Target. J. Med. Chem. 2011, 54, 26–53. [Google Scholar] [CrossRef] [PubMed]
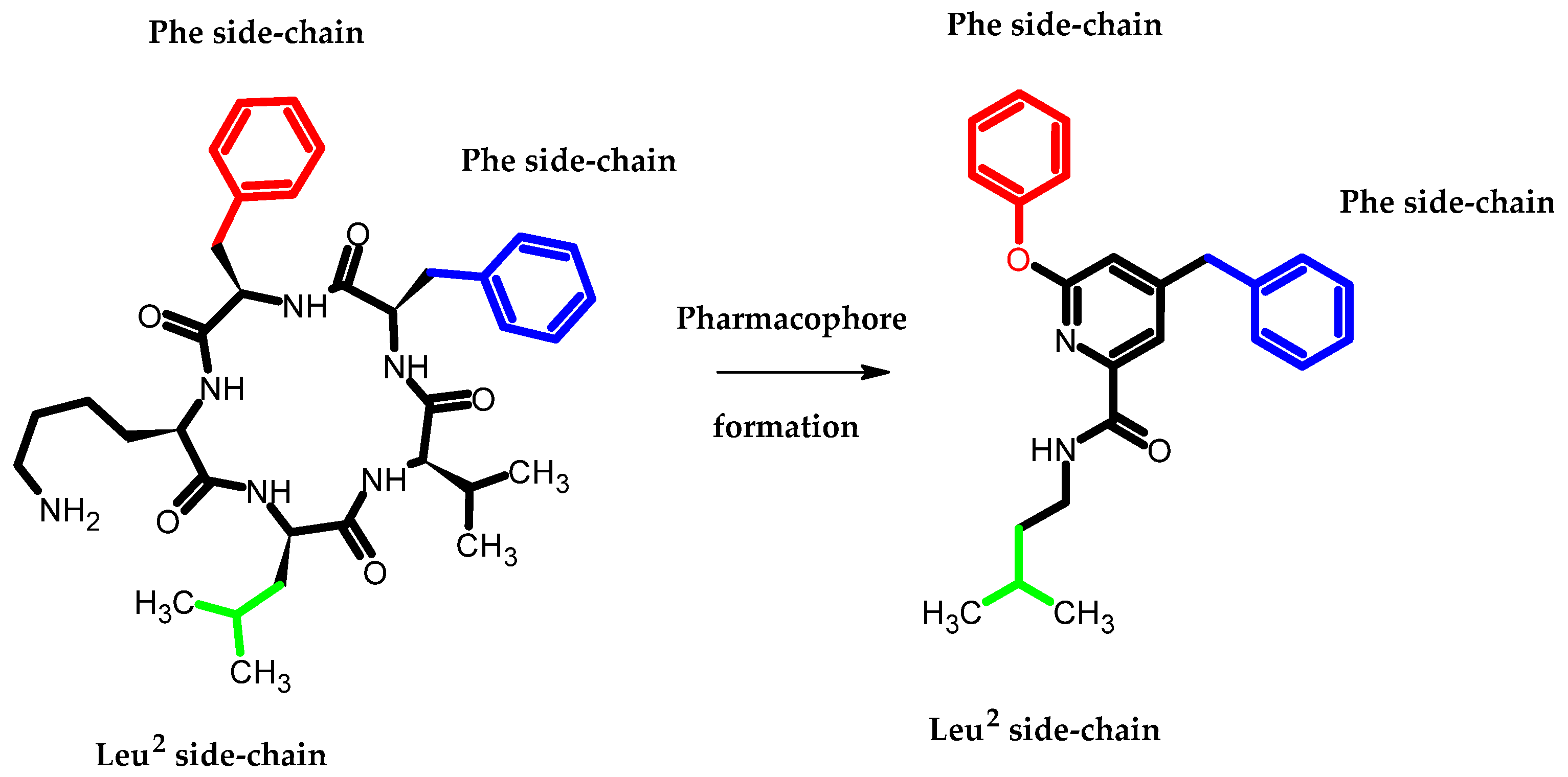
- Arai, T.; Araya, T.; Sasaki, D.; Taniguchi, A.; Sato, T.; Sohma, Y.; Kanai, M. Rational Design and Identification of a Non-Peptidic Aggregation Inhibitor of Amyloid-β Based on a Pharmacophore Motif Obtained from Cyclo[-Lys-Leu-Val-Phe-Phe-]. Angew. Chem. Int. Ed. 2014, 53, 8236–8239. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubacka, M.; Mogilski, S.; Bednarski, M.; Nowiński, L.; Dudek, M.; Żmudzka, E.; Siwek, A.; Waszkielewicz, A.M.; Marona, H.; Satała, G.; et al. Antidepressant-like Activity of Aroxyalkyl Derivatives of 2-Methoxyphenylpiperazine and Evidence for the Involvement of Serotonin Receptor Subtypes in Their Mechanism of Action. Pharmacol. Biochem. Behav. 2016, 141, 28–41. [Google Scholar] [CrossRef]
- Carr, G.V.; Schechter, L.E.; Lucki, I. Antidepressant and Anxiolytic Effects of Selective 5-HT6 Receptor Agonists in Rats. Psychopharmacology 2011, 213, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Artigas, F. Serotonin Receptors Involved in Antidepressant Effects. Pharmacol. Ther. 2013, 137, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhoury, M. Revisiting the Serotonin Hypothesis: Implications for Major Depressive Disorders. Mol. Neurobiol. 2016, 53, 2778–2786. [Google Scholar] [CrossRef]
- Waszkielewicz, A.M.; Pytka, K.; Rapacz, A.; Wełna, E.; Jarzyna, M.; Satała, G.; Bojarski, A.; Sapa, J.; Żmudzki, P.; Filipek, B.; et al. Synthesis and Evaluation of Antidepressant-like Activity of Some 4-Substituted 1-(2-Methoxyphenyl)Piperazine Derivatives. Chem. Biol. Drug Des. 2015, 85, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Del Bello, F.; Bonifazi, A.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Petrelli, R.; Cifani, C.; Micioni Di Bonaventura, M.V.; Keck, T.M.; Mazzolari, A.; et al. The Replacement of the 2-Methoxy Substituent of N-((6,6-Diphenyl-1,4-Dioxan-2-Yl)Methyl)-2-(2-Methoxyphenoxy)Ethan-1-Amine Improves the Selectivity for 5-HT1A Receptor over A1-Adrenoceptor and D2-like Receptor Subtypes. Eur. J. Med. Chem. 2017, 125, 233–244. [Google Scholar] [CrossRef]
- Szczepańska, K.; Karcz, T.; Mogilski, S.; Siwek, A.; Kuder, K.J.; Latacz, G.; Kubacka, M.; Hagenow, S.; Lubelska, A.; Olejarz, A.; et al. Synthesis and Biological Activity of Novel Tert-Butyl and Tert-Pentylphenoxyalkyl Piperazine Derivatives as Histamine H3R Ligands. Eur. J. Med. Chem. 2018, 152, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Hassan, A.H.E.; Jeong, H.; Kwon, Y.; Choi, J.G.; Oh, M.S.; Park, K.D.; Kim, Y.K.; Roh, E.J. First-in-Class DAPK1/CSF1R Dual Inhibitors: Discovery of 3,5-Dimethoxy-N-(4-(4-Methoxyphenoxy)-2-((6-Morpholinopyridin-3-Yl)Amino)Pyrimidin-5-Yl)Benzamide as a Potential Anti-Tauopathies Agent. Eur. J. Med. Chem. 2019, 162, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Franchini, S.; Sorbi, C.; Linciano, P.; Carnevale, G.; Tait, A.; Ronsisvalle, S.; Buccioni, M.; Del Bello, F.; Cilia, A.; Pirona, L.; et al. 1,3-Dioxane as a Scaffold for Potent and Selective 5-HT1AR Agonist with in-Vivo Anxiolytic, Anti-Depressant and Anti-Nociceptive Activity. Eur. J. Med. Chem. 2019, 176, 310–325. [Google Scholar] [CrossRef]
- Łażewska, D.; Jończyk, J.; Bajda, M.; Szałaj, N.; Więckowska, A.; Panek, D.; Moore, C.; Kuder, K.; Malawska, B.; Kieć-Kononowicz, K. Cholinesterase Inhibitory Activity of Chlorophenoxy Derivatives—Histamine H3 Receptor Ligands. Bioorg. Med. Chem. Lett. 2016, 26, 4140–4145. [Google Scholar] [CrossRef]
- Kaniakova, M.; Korabecny, J.; Holubova, K.; Kleteckova, L.; Chvojkova, M.; Hakenova, K.; Prchal, L.; Novak, M.; Dolezal, R.; Hepnarova, V.; et al. 7-Phenoxytacrine Is a Dually Acting Drug with Neuroprotective Efficacy In Vivo. Biochem. Pharmacol. 2021, 186, 114460. [Google Scholar] [CrossRef]
- Abatematteo, F.S.; Mosier, P.D.; Niso, M.; Brunetti, L.; Berardi, F.; Loiodice, F.; Contino, M.; Delprat, B.; Maurice, T.; Laghezza, A.; et al. Development of Novel Phenoxyalkylpiperidines as High-Affinity Sigma-1 (Σ1) Receptor Ligands with Potent Anti-Amnesic Effect. Eur. J. Med. Chem. 2022, 228, 114038. [Google Scholar] [CrossRef]
- Maurice, T.; Goguadze, N. Role of Σ1 Receptors in Learning and Memory and Alzheimer’s Disease-Type Dementia. Adv. Exp. Med. Biol. 2017, 964, 213–233. [Google Scholar] [CrossRef]
- Maurice, T.; Goguadze, N. Sigma-1 (Σ1) Receptor in Memory and Neurodegenerative Diseases. In Sigma Proteins: Evolution of the Concept of Sigma Receptors; Kim, F.J., Pasternak, G.W., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2017; pp. 81–108. ISBN 978-3-319-65853-7. [Google Scholar]
- Berardi, F.; Ferorelli, S.; Abate, C.; Pedone, M.P.; Colabufo, N.A.; Contino, M.; Perrone, R. Methyl Substitution on the Piperidine Ring of N-[ω-(6-Methoxynaphthalen-1-Yl)Alkyl] Derivatives as a Probe for Selective Binding and Activity at the Σ1 Receptor. J. Med. Chem. 2005, 48, 8237–8244. [Google Scholar] [CrossRef]
- Navidpour, L.; Shabani, S.; Heidari, A.; Bashiri, M.; Ebrahim-Habibi, A.; Shahhosseini, S.; Shafaroodi, H.; Abbas Tabatabai, S.; Toolabi, M. 5-[Aryloxypyridyl (or Nitrophenyl)]-4H-1,2,4-Triazoles as Novel Flexible Benzodiazepine Analogues: Synthesis, Receptor Binding Affinity and Lipophilicity-Dependent Anti-Seizure Onset of Action. Bioorg. Chem. 2021, 106, 104504. [Google Scholar] [CrossRef] [PubMed]
- Sternbach, L.H. The Benzodiazepine Story. J. Med. Chem. 1979, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA Receptor Signalling Mechanisms Revealed by Structural Pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef]
- Kuder, K.; Łażewska, D.; Latacz, G.; Schwed, J.S.; Karcz, T.; Stark, H.; Karolak-Wojciechowska, J.; Kieć-Kononowicz, K. Chlorophenoxy Aminoalkyl Derivatives as Histamine H3R Ligands and Antiseizure Agents. Bioorg. Med. Chem. 2016, 24, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 2022 Cancer Facts & Figures Cancer|Cancer Death Rate Drops. Available online: https://www.cancer.org/latest-news/facts-and-figures-2022.html (accessed on 29 March 2022).
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental Risk Factors for Cancer-Review Paper. Ann Agric Environ Med. 2019, 26, 1–7. [Google Scholar] [CrossRef]
- Hassan, R.A.; Emam, S.H.; Hwang, D.; Kim, G.-D.; Hassanin, S.O.; Khalil, M.G.; Abdou, A.M.; Sonousi, A. Design, Synthesis and Evaluation of Anticancer Activity of New Pyrazoline Derivatives by down-Regulation of VEGF: Molecular Docking and Apoptosis Inducing Activity. Bioorg. Chem. 2022, 118, 105487. [Google Scholar] [CrossRef]
- Glade-Bender, J.; Kandel, J.J.; Yamashiro, D.J. VEGF Blocking Therapy in the Treatment of Cancer. Expert Opin. Biol. Ther. 2003, 3, 263–276. [Google Scholar] [CrossRef]
- Prager, G.W.; Poettler, M.; Unseld, M.; Zielinski, C.C. Angiogenesis in Cancer: Anti-VEGF Escape Mechanisms. Transl. Lung Cancer Res. 2012, 1, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 Activity Up-Regulates VEGF Expression and Tumor Angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical Antiangiogenesis and Antitumor Activities of Axitinib (AG-013736), an Oral, Potent, and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palakhachane, S.; Ketkaew, Y.; Chuaypen, N.; Sirirak, J.; Boonsombat, J.; Ruchirawat, S.; Tangkijvanich, P.; Suksamrarn, A.; Limpachayaporn, P. Synthesis of Sorafenib Analogues Incorporating a 1,2,3-Triazole Ring and Cytotoxicity towards Hepatocellular Carcinoma Cell Lines. Bioorg. Chem. 2021, 112, 104831. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Sun, D.; Shi, D.; Wang, G.; Chen, Y.; Zhang, K.; Tan, H.; Liu, J.; Liu, B.; Ouyang, L. Design, Synthesis, and Biological Evaluation of Quinazolin-4(3H)-One Derivatives Co-Targeting Poly(ADP-Ribose) Polymerase-1 and Bromodomain Containing Protein 4 for Breast Cancer Therapy. Acta Pharm. Sin. B 2021, 11, 156–180. [Google Scholar] [CrossRef]
- Güngör, T.; Ozleyen, A.; Yılmaz, Y.B.; Siyah, P.; Ay, M.; Durdağı, S.; Tumer, T.B. New Nimesulide Derivatives with Amide/Sulfonamide Moieties: Selective COX-2 Inhibition and Antitumor Effects. Eur. J. Med. Chem. 2021, 221, 113566. [Google Scholar] [CrossRef]
- Li, F.; Li, X.-M.; Sheng, D.; Chen, S.-R.; Nie, X.; Liu, Z.; Wang, D.; Zhao, Q.; Wang, Y.; Wang, Y.; et al. Discovery and Preliminary SAR of 14-Aryloxy-Andrographolide Derivatives as Antibacterial Agents with Immunosuppressant Activity. RSC Adv. 2018, 8, 9440–9456. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, F.; Tang, F.; Zhang, J.; Li, R.; Sheng, D.; Lee, S.M.-Y.; Zhou, G.-C.; Leung, G.P.-H. AGS-30, an Andrographolide Derivative, Suppresses Tumor Angiogenesis and Growth in Vitro and in Vivo. Biochem. Pharmacol. 2020, 171, 113694. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, J.; Wei, X.; Pei, Y.; Ye, J.; Li, X.; Si, G.; Tian, J.; Dong, Y.; Liu, G. Nonpeptidic Quinazolinone Derivatives as Dual Nucleotide-Binding Oligomerization Domain-like Receptor 1/2 Antagonists for Adjuvant Cancer Chemotherapy. Eur. J. Med. Chem. 2020, 207, 112723. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.H.; Ferrero, R.L.; Philpott, D.J.; Girardin, S.E. Nod-like Proteins in Immunity, Inflammation and Disease. Nat. Immunol. 2006, 7, 1250–1257. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in Innate Immunity and Inflammatory Diseases. Biosci. Rep. 2012, 32, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Miceli-Richard, C.; Lesage, S.; Rybojad, M.; Prieur, A.M.; Manouvrier-Hanu, S.; Häfner, R.; Chamaillard, M.; Zouali, H.; Thomas, G.; Hugot, J.P. CARD15 Mutations in Blau Syndrome. Nat. Genet. 2001, 29, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A Frameshift Mutation in NOD2 Associated with Susceptibility to Crohn’s Disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Xu, L.; Hong, D.; Zhang, X.; Liu, J.; Li, D.; Li, J.; Zhou, Y.; Liu, T. Design, Synthesis, and Biological Evaluation of Novel Phenol Ether Derivatives as Non-Covalent Proteasome Inhibitors. Eur. J. Med. Chem. 2019, 161, 543–558. [Google Scholar] [CrossRef] [PubMed]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How Proteolysis Drives the Cell Cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef]
- Ciechanover, A. The Ubiquitin-Proteasome Pathway: On Protein Death and Cell Life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-J.; Lin, F.; Qin, Z.-H. The Roles of the Proteasome Pathway in Signal Transduction and Neurodegenerative Diseases. Neurosci. Bull. 2008, 24, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A Selective Inhibitor of the Immunoproteasome Subunit LMP7 Blocks Cytokine Production and Attenuates Progression of Experimental Arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Lakshmithendral, K.; Saravanan, K.; Elancheran, R.; Archana, K.; Manikandan, N.; Arjun, H.A.; Ramanathan, M.; Lokanath, N.K.; Kabilan, S. Design, Synthesis and Biological Evaluation of 2-(Phenoxymethyl)-5-Phenyl-1,3,4-Oxadiazole Derivatives as Anti-Breast Cancer Agents. Eur. J. Med. Chem. 2019, 168, 1–10. [Google Scholar] [CrossRef]
- Mohammed, Y.H.E.; Malojirao, V.H.; Thirusangu, P.; Al-Ghorbani, M.; Prabhakar, B.T.; Khanum, S.A. The Novel 4-Phenyl-2-Phenoxyacetamide Thiazoles Modulates the Tumor Hypoxia Leading to the Crackdown of Neoangiogenesis and Evoking the Cell Death. Eur. J. Med. Chem. 2018, 143, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Milik, S.N.; Abdel-Aziz, A.K.; Lasheen, D.S.; Serya, R.A.T.; Minucci, S.; Abouzid, K.A.M. Surmounting the Resistance against EGFR Inhibitors through the Development of Thieno [2,3-d]Pyrimidine-Based Dual EGFR/HER2 Inhibitors. Eur. J. Med. Chem. 2018, 155, 316–336. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic Kinase Signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citri, A.; Yarden, Y. EGF–ERBB Signalling: Towards the Systems Level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB Receptors and Cancer: The Complexity of Targeted Inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Rowinsky, E.K. The ErbB Family: Targets for Therapeutic Development against Cancer and Therapeutic Strategies Using Monoclonal Antibodies and Tyrosine Kinase Inhibitors. Annu. Rev. Med. 2004, 55, 433–457. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor-Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Bianco, R.; Gelardi, T.; Damiano, V.; Ciardiello, F.; Tortora, G. Rational Bases for the Development of EGFR Inhibitors for Cancer Treatment. Int. J. Biochem. Cell Biology 2007, 39, 1416–1431. [Google Scholar] [CrossRef]
- Kamath, S.; Buolamwini, J.K. Targeting EGFR and HER-2 Receptor Tyrosine Kinases for Cancer Drug Discovery and Development. Med. Res. Rev. 2006, 26, 569–594. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, Y.H.E.; Thirusangu, P.; Zabiulla; Vigneshwaran, V.; Prabhakar, B.T.; Khanum, S.A. The Anti-Invasive Role of Novel Synthesized Pyridazine Hydrazide Appended Phenoxy Acetic Acid against Neoplastic Development Targeting Matrix Metallo Proteases. Biomed. Pharmacother. 2017, 95, 375–386. [Google Scholar] [CrossRef]
- Xie, R.; Yao, Y.; Tang, P.; Chen, G.; Liu, X.; Yun, F.; Cheng, C.; Wu, X.; Yuan, Q. Design, Synthesis and Biological Evaluation of Novel Hydroxamates and 2-Aminobenzamides as Potent Histone Deacetylase Inhibitors and Antitumor Agents. Eur. J. Med. Chem. 2017, 134, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulabaş, N.; Tatar, E.; Bingöl Özakpınar, Ö.; Özsavcı, D.; Pannecouque, C.; De Clercq, E.; Küçükgüzel, İ. Synthesis and Antiproliferative Evaluation of Novel 2-(4H-1,2,4-Triazole-3-Ylthio)Acetamide Derivatives as Inducers of Apoptosis in Cancer Cells. Eur. J. Med. Chem. 2016, 121, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Pingaew, R.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Design, Synthesis and Molecular Docking Studies of Novel N-Benzenesulfonyl-1,2,3,4-Tetrahydroisoquinoline-Based Triazoles with Potential Anticancer Activity. Eur. J. Med. Chem. 2014, 81, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Sen Gupta, A.K.; Misra, H.K. Studies on potential pesticides. Part XIII: Synthesis and evaluation of S-(3-substituted phenoxymethyl-4-aryl/ayclohexyl-4H-1,2,4-triazol-5-yl)-2-mercaptomethylbenzimidazo-les for antibacterial and insecticidal activities. J. Indian Chem. Soc. 1981, 58, 508–511. [Google Scholar]
- Pitucha, M.; Korga-Plewko, A.; Kozyra, P.; Iwan, M.; Kaczor, A.A. 2,4-Dichlorophenoxyacetic Thiosemicarbazides as a New Class of Compounds against Stomach Cancer Potentially Intercalating with DNA. Biomolecules 2020, 10, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyra, P.; Korga-Plewko, A.; Karczmarzyk, Z.; Hawrył, A.; Wysocki, W.; Człapski, M.; Iwan, M.; Ostrowska-Leśko, M.; Fornal, E.; Pitucha, M. Potential Anticancer Agents against Melanoma Cells Based on an As-Synthesized Thiosemicarbazide Derivative. Biomolecules 2022, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and Cancer: Promising Prospects to Be Explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef]
- Kozyra, P.; Krasowska, D.; Pitucha, M. New Potential Agents for Malignant Melanoma Treatment—Most Recent Studies 2020–2022. Int. J. Mol. Sci. 2022, 23, 6084. [Google Scholar] [CrossRef]
- Burger, J.A. BTK Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Schnute, M.E.; Benoit, S.E.; Buchler, I.P.; Caspers, N.; Grapperhaus, M.L.; Han, S.; Hotchandani, R.; Huang, N.; Hughes, R.O.; Juba, B.M.; et al. Aminopyrazole Carboxamide Bruton’s Tyrosine Kinase Inhibitors. Irreversible to Reversible Covalent Reactive Group Tuning. ACS Med. Chem. Lett. 2019, 10, 80–85. [Google Scholar] [CrossRef]
- Zhang, C.; Pei, H.; He, J.; Zhu, J.; Li, W.; Niu, T.; Xiang, M.; Chen, L. Design, Synthesis and Evaluation of Novel 7H-Pyrrolo[2,3-d]Pyrimidin-4-Amine Derivatives as Potent, Selective and Reversible Bruton’s Tyrosine Kinase (BTK) Inhibitors for the Treatment of Rheumatoid Arthritis. Eur. J. Med. Chem. 2019, 169, 121–143. [Google Scholar] [CrossRef]
- Zheng, N.; Pan, J.; Hao, Q.; Li, Y.; Zhou, W. Design, Synthesis and Biological Evaluation of Novel 3-Substituted Pyrazolopyrimidine Derivatives as Potent Bruton’s Tyrosine Kinase (BTK) Inhibitors. Bioorg. Med. Chem. 2018, 26, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, S.; Zhang, Z.; Zhang, C.; Zeng, S.; Liang, M.; Shen, Z.; Liu, X. HZ-A-005, a Potent, Selective, and Covalent Bruton’s Tyrosine Kinase Inhibitor in Preclinical Development. Bioorg. Chem. 2020, 105, 104377. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Liu-Bujalski, L.; Caldwell, R.D.; Follis, A.V.; Gardberg, A.; Goutopoulos, A.; Grenningloh, R.; Head, J.; Johnson, T.; Jones, R.; et al. Discovery of Potent, Highly Selective Covalent Irreversible BTK Inhibitors from a Fragment Hit. Bioorg. Med. Chem. Lett. 2018, 28, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-Generation Inhibitors of Bruton Tyrosine Kinase. J. Hematol. Oncol. 2016, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Norman, P. Investigational Bruton’s Tyrosine Kinase Inhibitors for the Treatment of Rheumatoid Arthritis. Expert Opin. Investig. Drugs 2016, 25, 891–899. [Google Scholar] [CrossRef]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef]
- Lou, Y.; Owens, T.D.; Kuglstatter, A.; Kondru, R.K.; Goldstein, D.M. Bruton’s Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evaluation for Inflammatory Diseases and B Cell Malignancies. J. Med. Chem. 2012, 55, 4539–4550. [Google Scholar] [CrossRef]
- CDC What Exactly Is Antibiotic Resistance? Available online: https://www.cdc.gov/drugresistance/about.html (accessed on 29 March 2022).
- Castelino, P.A.; Naik, P.; Dasappa, J.P.; Sujayraj, R.S.; Sharath Chandra, K.; Chaluvaiah, K.; Nair, R.; Sandya Kumari, M.V.; Kalthur, G.; Adiga, S.K. Synthesis of Novel Thiadiazolotriazin-4-Ones and Study of Their Mosquito-Larvicidal and Antibacterial Properties. Eur. J. Med. Chem. 2014, 84, 194–199. [Google Scholar] [CrossRef]
- Basanagouda, M.; Jambagi, V.B.; Barigidad, N.N.; Laxmeshwar, S.S.; Devaru, V. Narayanachar, null Synthesis, Structure-Activity Relationship of Iodinated-4-Aryloxymethyl-Coumarins as Potential Anti-Cancer and Anti-Mycobacterial Agents. Eur. J. Med. Chem. 2014, 74, 225–233. [Google Scholar] [CrossRef]
- Karad, S.C.; Purohit, V.B.; Raval, D.K. Design, Synthesis and Characterization of Fluoro Substituted Novel Pyrazolylpyrazolines Scaffold and Their Pharmacological Screening. Eur. J. Med. Chem. 2014, 84, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; De Giuli Morghen, C.; et al. Benzodioxane-Benzamides as New Bacterial Cell Division Inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Kanetaka, H.; Koseki, Y.; Taira, J.; Umei, T.; Komatsu, H.; Sakamoto, H.; Gulten, G.; Sacchettini, J.C.; Kitamura, M.; Aoki, S. Discovery of InhA Inhibitors with Anti-Mycobacterial Activity through a Matched Molecular Pair Approach. Eur. J. Med. Chem. 2015, 94, 378–385. [Google Scholar] [CrossRef]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to Synthesis and Processing of Mycolic Acids in Mycobacterium Tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Kim, Y.M.; Kim, R.Y.; Seo, M.J.; No, Z.; Nam, K.; Kim, S.; Kim, J. Synthesis and Structure-Activity Studies of Side Chain Analogues of the Anti-Tubercular Agent, Q203. Eur. J. Med. Chem. 2017, 125, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Muğlu, H.; Şener, N.; Mohammad Emsaed, H.A.; Özkınalı, S.; Özkan, O.E.; Gür, M. Synthesis and Characterization of 1,3,4-Thiadiazole Compounds Derived from 4-Phenoxybutyric Acid for Antimicrobial Activities. J. Mol. Struct. 2018, 1174, 151–159. [Google Scholar] [CrossRef]
- D’Souza, V.T.; Nayak, J.; D’Mello, D.E.; Dayananda, P. Synthesis and Characterization of Biologically Important Quinoline Incorporated Triazole Derivatives. J. Mol. Struct. 2021, 1229, 129503. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule Vikas, D.; Naveen; Lal, K. Synthesis, Antifungal Studies, Molecular Docking, ADME and DNA Interaction Studies of 4-Hydroxyphenyl Benzothiazole Linked 1,2,3-Triazoles. J. Mol. Struct. 2021, 1245, 131013. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, Y.; Li, L.; Zhao, R.; Wang, B.; Lv, K.; Liu, M.; You, X. Identification of N-(2-Phenoxyethyl)Imidazo[1,2-a]Pyridine-3-Carboxamides as New Antituberculosis Agents. ACS Med. Chem. Lett. 2016, 7, 1130–1133. [Google Scholar] [CrossRef] [Green Version]
- Global HIV & AIDS Statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 30 March 2022).
- Wang, J.; Zhan, P.; Li, Z.; Liu, H.; De Clercq, E.; Pannecouque, C.; Liu, X. Discovery of Nitropyridine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors via a Structure-Based Core Refining Approach. Eur. J. Med. Chem. 2014, 76, 531–538. [Google Scholar] [CrossRef]
- Singh, A.K.; Das, K. Insights into HIV-1 Reverse Transcriptase (RT) Inhibition and Drug Resistance from Thirty Years of Structural Studies. Viruses 2022, 14, 1027. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Tian, Y.; Liu, H.; Zhan, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J.; Liu, X. Discovery of Novel Diarylpyrimidines as Potent HIV NNRTIs via a Structure-Guided Core-Refining Approach. Eur. J. Med. Chem. 2014, 80, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Liang, X.; Li, C.; Chen, W.; Liu, T.; Li, X.; Sun, Y.; Fu, L.; Liu, H.; De Clercq, E.; et al. Fused Heterocycles Bearing Bridgehead Nitrogen as Potent HIV-1 NNRTIs. Part 4: Design, Synthesis and Biological Evaluation of Novel Imidazo[1,2-a]Pyrazines. Eur. J. Med. Chem. 2015, 93, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Chen, X.; Kang, D.; Huang, B.; Li, W.; Zhan, P.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Liu, X. Design, Synthesis and Evaluation of Novel HIV-1 NNRTIs with Dual Structural Conformations Targeting the Entrance Channel of the NNRTI Binding Pocket. Eur J Med Chem 2016, 115, 53–62. [Google Scholar] [CrossRef]
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The Past and the Present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; Van Den Abbeele, J.; et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 2016, 19, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainy, J.; Sharma, R. Synthesis, Antimalarial Evaluation and Molecular Docking Studies of Some Thiolactone Derivatives. J. Mol. Struct. 2017, 1134, 350–359. [Google Scholar] [CrossRef]
- Otero, E.; García, E.; Palacios, G.; Yepes, L.M.; Carda, M.; Agut, R.; Vélez, I.D.; Cardona, W.I.; Robledo, S.M. Triclosan-Caffeic Acid Hybrids: Synthesis, Leishmanicidal, Trypanocidal and Cytotoxic Activities. Eur. J. Med. Chem. 2017, 141, 73–83. [Google Scholar] [CrossRef]
- López-Lira, C.; Tapia, R.A.; Herrera, A.; Lapier, M.; Maya, J.D.; Soto-Delgado, J.; Oliver, A.G.; Graham Lappin, A.; Uriarte, E. New Benzimidazolequinones as Trypanosomicidal Agents. Bioorg. Chem. 2021, 111, 104823. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Bergamini, C.; Molina, M.T.; Falchi, F.; Cavalli, A.; Kaiser, M.; Brun, R.; Fato, R.; Bolognesi, M.L. 2-Phenoxy-1,4-Naphthoquinones: From a Multitarget Antitrypanosomal to a Potential Antitumor Profile. J. Med. Chem. 2015, 58, 6422–6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijma, H.J.; Groeneveld, G.J. Analgesic Drug Development: Proof-of-Mechanism and Proof-of-Concept in Early Phase Clinical Studies. Med. Drug Discov. 2021, 10, 100083. [Google Scholar] [CrossRef]
- Farag, A.K.; Elkamhawy, A.; Londhe, A.M.; Lee, K.-T.; Pae, A.N.; Roh, E.J. Novel LCK/FMS Inhibitors Based on Phenoxypyrimidine Scaffold as Potential Treatment for Inflammatory Disorders. Eur. J. Med. Chem. 2017, 141, 657–675. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, H.M.; Al-Ostoot, F.H.; Vivek, H.K.; Khanum, S.A. Design, Docking, Synthesis, and Characterization of Novel N’(2-Phenoxyacetyl) Nicotinohydrazide and N’(2-Phenoxyacetyl)Isonicotinohydrazide Derivatives as Anti-Inflammatory and Analgesic Agents. J. Mol. Struct. 2022, 1247, 131404. [Google Scholar] [CrossRef]
- Gunaydin, C.; Bilge, S.S. Effects of Nonsteroidal Anti-Inflammatory Drugs at the Molecular Level. Eurasian J. Med. 2018, 50, 116–121. [Google Scholar] [CrossRef]
- Carrasco, E.; Gomez-Gutierrez, P.; Campos, P.M.; Vega, M.; Messeguer, A.; Perez, J.J. Discovery of Novel 2,3,5-Trisubstituted Pyridine Analogs as Potent Inhibitors of IL-1β via Modulation of the P38 MAPK Signaling Pathway. Eur. J. Med. Chem. 2021, 223, 113620. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliorini, P.; Italiani, P.; Pratesi, F.; Puxeddu, I.; Boraschi, D. The IL-1 Family Cytokines and Receptors in Autoimmune Diseases. Autoimmun. Rev. 2020, 19, 102617. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ostoot, F.H.; Zabiulla; Grisha, S.; Mohammed, Y.H.E.; Vivek, H.K.; Ara Khanum, S. Molecular Docking and Synthesis of Caffeic Acid Analogous and Its Anti-Inflammatory, Analgesic and Ulcerogenic Studies. Bioorg. Med. Chem. Lett. 2021, 33, 127743. [Google Scholar] [CrossRef]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-Induced Edema in Hind Paw of the Rat as an Assay for Antiinflammatory Drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, H.; Hanekamp, W.; Lehr, M. (Indolylalkyl)Piperidine Carbamates as Inhibitors of Fatty Acid Amide Hydrolase (FAAH). Med. Chem. Commun. 2017, 8, 616–620. [Google Scholar] [CrossRef]
- Keith, J.M.; Tichenor, M.S.; Apodaca, R.L.; Xiao, W.; Jones, W.M.; Seierstad, M.; Pierce, J.M.; Palmer, J.A.; Webb, M.; Karbarz, M.J.; et al. The SAR of Brain Penetration for a Series of Heteroaryl Urea FAAH Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3109–3114. [Google Scholar] [CrossRef]
- Sundermann, T.; Hanekamp, W.; Lehr, M. Structure–Activity Relationship Studies on 1-Heteroaryl-3-Phenoxypropan-2-Ones Acting as Inhibitors of Cytosolic Phospholipase A2α and Fatty Acid Amide Hydrolase: Replacement of the Activated Ketone Group by Other Serine Traps. J. Enzym. Inhib. Med. Chem. 2016, 31, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Althaus, J.; Hake, T.; Hanekamp, W.; Lehr, M. 1-(5-Carboxyindazol-1-Yl)Propan-2-Ones as Dual Inhibitors of Cytosolic Phospholipase A2α and Fatty Acid Amide Hydrolase: Bioisosteric Replacement of the Carboxylic Acid Moiety. J. Enzym. Inhib. Med. Chem. 2016, 31, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharroubi, A.T.; Darwish, H.M. Diabetes Mellitus: The Epidemic of the Century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Diabetes. Available online: https://www.who.int/westernpacific/health-topics/diabetes (accessed on 2 April 2022).
- Desai, J.; Patel, B.; Darji, B.; Gite, A.; Panchal, N.; Bhosale, G.; Shedage, S.; Patel, S.; Kadam, P.; Patel, G.; et al. Discovery of Novel, Potent and Orally Efficacious Inhibitor of Neutral Amino Acid Transporter B0AT1 (SLC6A19). Bioorg. Med. Chem. Lett. 2021, 53, 128421. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Klingel, K.; Kowalczuk, S.; Rasko, J.E.J.; Cavanaugh, J.; Bröer, S. Molecular Cloning of Mouse Amino Acid Transport System B0, a Neutral Amino Acid Transporter Related to Hartnup Disorder. J. Biol. Chem. 2004, 279, 24467–24476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Rose, A.J.; Sijmonsma, T.P.; Bröer, A.; Pfenninger, A.; Herzig, S.; Schmoll, D.; Bröer, S. Mice Lacking Neutral Amino Acid Transporter B(0)AT1 (Slc6a19) Have Elevated Levels of FGF21 and GLP-1 and Improved Glycaemic Control. Mol. Metab. 2015, 4, 406–417. [Google Scholar] [CrossRef]
- Cheng, Q.; Shah, N.; Bröer, A.; Fairweather, S.; Jiang, Y.; Schmoll, D.; Corry, B.; Bröer, S. Identification of Novel Inhibitors of the Amino Acid Transporter B0 AT1 (SLC6A19), a Potential Target to Induce Protein Restriction and to Treat Type 2 Diabetes. Br. J. Pharmacol. 2017, 174, 468–482. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; You, Y.; Li, H.; Cheng, Y.; Qian, M.; Zhou, X.; Yuan, H.; Xu, Q.-L.; Dai, L.; Wang, P.; et al. Discovery of AdipoRon Analogues as Novel AMPK Activators without Inhibiting Mitochondrial Complex I. Eur. J. Med. Chem. 2020, 200, 112466. [Google Scholar] [CrossRef]
- Deshpande, A.M.; Bhuniya, D.; De, S.; Dave, B.; Vyavahare, V.P.; Kurhade, S.H.; Kandalkar, S.R.; Naik, K.P.; Kobal, B.S.; Kaduskar, R.D.; et al. Discovery of Liver-Directed Glucokinase Activator Having Anti-Hyperglycemic Effect without Hypoglycemia. Eur. J. Med. Chem. 2017, 133, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual Roles for Glucokinase in Glucose Homeostasis as Determined by Liver and Pancreatic Beta Cell-Specific Gene Knock-Outs Using Cre Recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, N.; Farrelly, D.; Hagan, D.; Hillyer, D.; Arbeeny, C.; Sabrah, T.; Treloar, A.; Brown, K.; Kalinowski, S.; Mookhtiar, K. Expression of Human Hepatic Glucokinase in Transgenic Mice Liver Results in Decreased Glucose Levels and Reduced Body Weight. Diabetes 1997, 46, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Jackerott, M.; Baudry, A.; Bucchini, D.; Jami, J.; Joshi, R. Improved Metabolic Disorders of Insulin Receptor-Deficient Mice by Transgenic Overexpression of Glucokinase in the Liver. Diabetologia 2002, 45, 1292–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, T.P.; Catlin, R.L.; Chan, R.; Fujimoto, Y.; Sasaki, N.; Printz, R.L.; Newgard, C.B.; Shiota, M. Restoration of Hepatic Glucokinase Expression Corrects Hepatic Glucose Flux and Normalizes Plasma Glucose in Zucker Diabetic Fatty Rats. Diabetes 2009, 58, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Q.; Xu, Q.; Luo, J.; Wang, L.-J.; Jiang, B.; Zhang, R.-S.; Shi, D.-Y. Design, Synthesis and Biological Evaluation of Uncharged Catechol Derivatives as Selective Inhibitors of PTP1B. Eur. J. Med. Chem. 2017, 136, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, C.-S.; Gao, L.-X.; Gong, J.-X.; Wang, Z.-H.; Li, J.-Y.; Li, J.; Li, X.-W.; Guo, Y.-W. Design, Synthesis and in Vitro Activity of Phidianidine B Derivatives as Novel PTP1B Inhibitors with Specific Selectivity. Bioorg. Med. Chem. Lett. 2016, 26, 778–781. [Google Scholar] [CrossRef]
- Goldstein, B.J. Protein-Tyrosine Phosphatase 1B (PTP1B): A Novel Therapeutic Target for Type 2 Diabetes Mellitus, Obesity and Related States of Insulin Resistance. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2001, 1, 265–275. [Google Scholar] [CrossRef]
- Comeau, A.B.; Critton, D.A.; Page, R.; Seto, C.T. A Focused Library of Protein Tyrosine Phosphatase Inhibitors. J. Med. Chem. 2010, 53, 6768–6772. [Google Scholar] [CrossRef]
- Kenner, K.A.; Anyanwu, E.; Olefsky, J.M.; Kusari, J. Protein-Tyrosine Phosphatase 1B Is a Negative Regulator of Insulin- and Insulin-like Growth Factor-I-Stimulated Signaling. J. Biol. Chem. 1996, 271, 19810–19816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, J.B.P.; do AF Navarro, D.M.; da Silva, A.G.; Santos, G.K.; Dutra, K.A.; Moreira, D.R.; Ramos, M.N.; Espíndola, J.W.P.; de Oliveira, A.D.T.; Brondani, D.J.; et al. Thiosemicarbazones as Aedes Aegypti Larvicidal. Eur. J. Med. Chem. 2015, 100, 162–175. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, Y.; Hu, L. Identification and Synthesis of Selective Cholesterol Esterase Inhibitor Using Dynamic Combinatorial Chemistry. Bioorg. Chem. 2022, 119, 105520. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhao, L.; Wang, L.; Chen, H.; Qiu, Y.; Wang, J.; Yang, H.; Liu, J.; Liu, H. Design, Synthesis, and Biological Evaluation of 2-(Phenoxyaryl)-3-Urea Derivatives as Novel P2Y1 Receptor Antagonists. Eur. J. Med. Chem. 2018, 158, 302–310. [Google Scholar] [CrossRef]
- Liu, E.C.-K.; Abell, L.M. Development and Validation of a Platelet Calcium Flux Assay Using a Fluorescent Imaging Plate Reader. Anal. Biochem. 2006, 357, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Sakauchi, N.; Furukawa, H.; Shirai, J.; Sato, A.; Kuno, H.; Saikawa, R.; Yoshida, M. Identification of 3,4-Dihydro-2H-Thiochromene 1,1-Dioxide Derivatives with a Phenoxyethylamine Group as Highly Potent and Selective A1D Adrenoceptor Antagonists. Eur. J. Med. Chem. 2017, 139, 114–127. [Google Scholar] [CrossRef] [PubMed]


























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozyra, P.; Pitucha, M. Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022. Int. J. Mol. Sci. 2022, 23, 8874. https://doi.org/10.3390/ijms23168874
Kozyra P, Pitucha M. Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022. International Journal of Molecular Sciences. 2022; 23(16):8874. https://doi.org/10.3390/ijms23168874
Chicago/Turabian StyleKozyra, Paweł, and Monika Pitucha. 2022. "Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022" International Journal of Molecular Sciences 23, no. 16: 8874. https://doi.org/10.3390/ijms23168874
APA StyleKozyra, P., & Pitucha, M. (2022). Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022. International Journal of Molecular Sciences, 23(16), 8874. https://doi.org/10.3390/ijms23168874