
Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder

 , , , , , and add
Show full author list
, , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
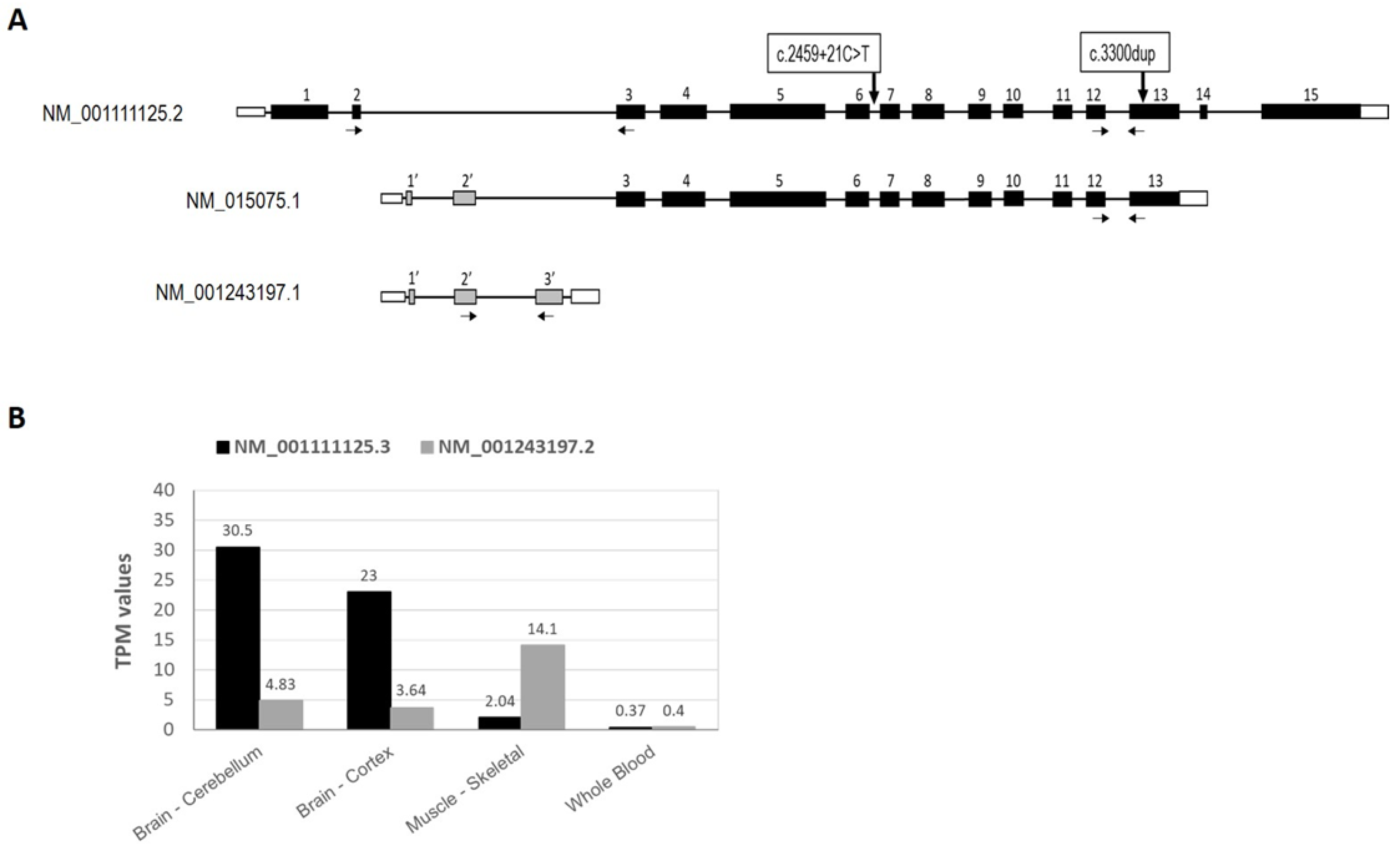
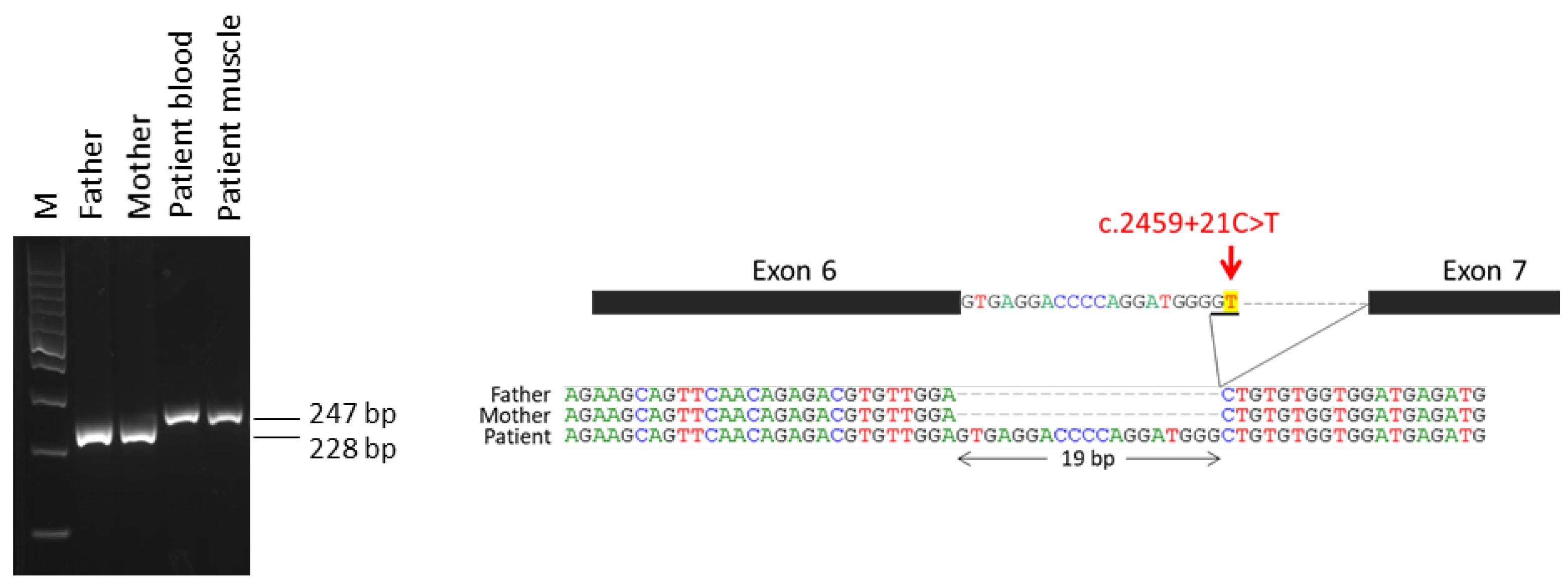
2.1. Variants in IQSEC2 Gene in the Patients
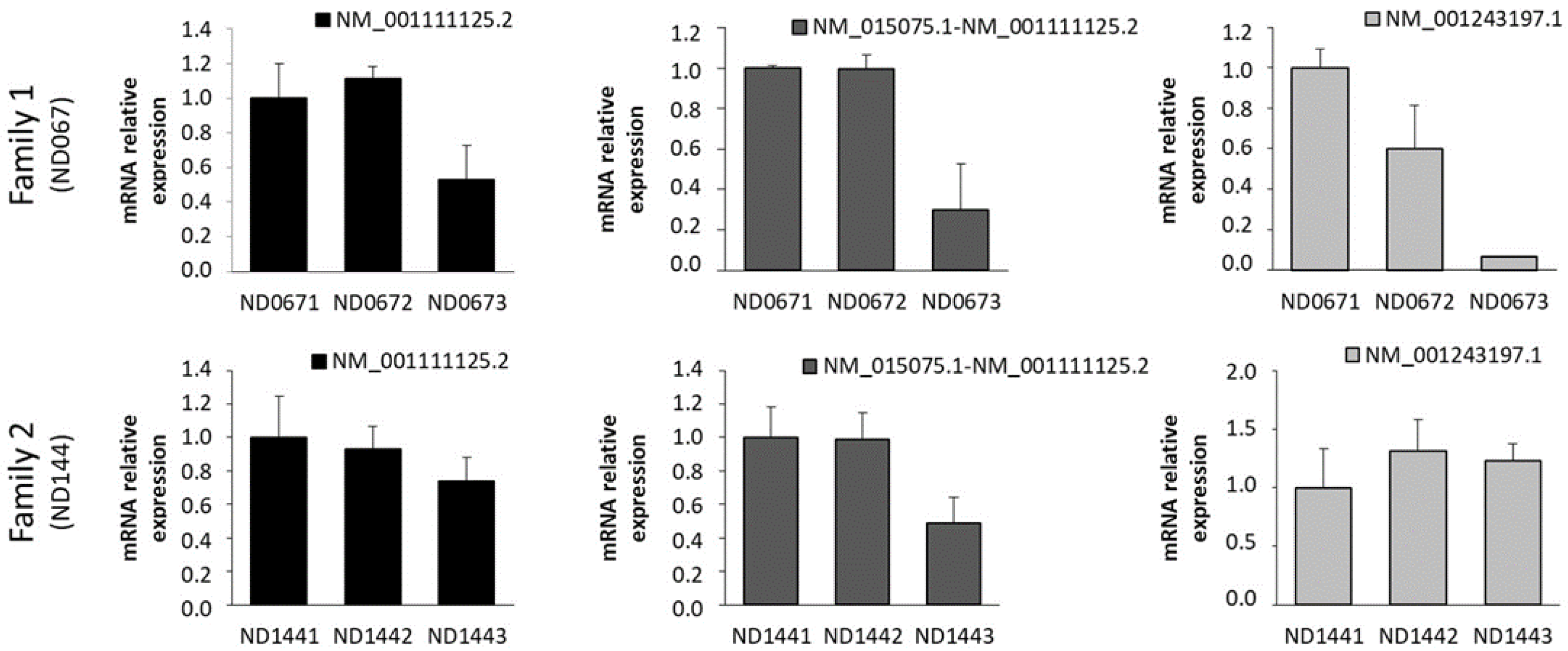
2.2. Expression of IQSEC2 Long and Short Transcript Isoforms by QT-PCR
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Trio Exome Analyses
4.3. IQSEC2 Gene Expression and Isoform Quantification
4.4. In Silico Prediction of Transcription Factor Binding
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, N.S.; Umanah, G.K.E.; Rogers, E.J.; Jada, R.; Lache, O.; Levy, A.P. IQSEC2-Associated Intellectual Disability and Autism. Int. J. Mol. Sci. 2019, 20, 3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoubridge, C.; Walikonis, R.S.; Gécz, J.; Harvey, R.J. Subtle functional defects in the Arf-specific guanine nucleotide exchange factor IQSEC2 cause non-syndromic X-linked intellectual disability. Small GTPases 2010, 1, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignot, C.; McMahon, A.C.; Bar, C.; Campeau, P.M.; Davidson, C.; Buratti, J.; Nava, C.; Jacquemont, M.L.; Tallot, M.; Milh, M.; et al. Correction: IQSEC2-related en-cephalopathy in males and females: A comparative study including 37 novel patients. Genet. Med. 2019, 21, 1897–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerem, A.; Haginoya, K.; Lev, D.; Blumkin, L.; Kivity, S.; Linder, I.; Shoubridge, C.; Palmer, E.E.; Field, M.; Boyle, J.; et al. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia 2016, 57, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, C.; Harvey, R.J.; Dudding-Byth, T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum. Mutat. 2019, 40, 5–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran Mau-Them, F.; Willems, M.; Albrecht, B.; Sanchez, E.; Puechberty, J.; Endele, S.; Schneider, A.; Ruiz Pallares, N.; Missirian, C.; Rivier, F.; et al. Expanding the phenotype of IQSEC2 mutations: Truncating mutations in severe intellectual disability. Eur. J. Hum. Genet. 2014, 22, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, H.; Sanda, M.; Fukaya, M.; Miyazaki, T.; Sukegawa, J.; Yanagisawa, T.; Suzuki, T.; Fukunaga, K.; Watanabe, M.; Kondo, H. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci. Res. 2008, 60, 199–212. [Google Scholar] [CrossRef]
- Radley, J.A.; O’Sullivan, R.B.G.; Turton, S.E.; Cox, H.; Vogt, J.; Morton, J.; Jones, E.; Smithson, S.; Lachlan, K.; Rankin, J.; et al. Deep phenotyping of 14 new patients with IQSEC2 variants, including monozygotic twins of discordant phenotype. Clin. Genet. 2019, 95, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.E. Regulation of Arf activation: The Sec7 family of guanine nucleotide exchange factors. Traffic 2007, 8, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Sanda, M.; Kamata, A.; Katsumata, O.; Fukunaga, K.; Watanabe, M.; Kondo, H.; Sakagami, H. The postsynaptic density protein, IQ-ArfGEF/BRAG1, can interact with IRSp53 through its proline-rich sequence. Brain Res. 2009, 1251, 7–15. [Google Scholar] [CrossRef] [PubMed]
- López-Martín, E.; Martínez-Delgado, B.; Bermejo-Sánchez, E.; Alonso, J.; Posada, M. SpainUDP: The Spanish Undiag-nosed Rare Diseases Program. Int. J. Environ. Res. Public Health 2018, 15, 1746. [Google Scholar] [CrossRef] [Green Version]
- Taruscio, D.; Groft, S.C.; Cederroth, H.; Melegh, B.; Lasko, P.; Kosaki, K.; Baynam, G.; McCray, A.; Gahl, W.A. Undiagnosed Diseases Network International (UDNI): White paper for global actions to meet patient needs. Mol. Genet. Metab. 2015, 116, 223–225. [Google Scholar] [CrossRef] [Green Version]
- Madrigal, I.; Alvarez-Mora, M.I.; Rosell, J.; Rodríguez-Revenga, L.; Karlberg, O.; Sauer, S.; Syvänen, A.C.; Mila, M. A novel splicing mutation in the IQSEC2 gene that modulates the phenotype severity in a family with intellectual disability. Eur. J. Hum. Genet. 2016, 24, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Lopergolo, D.; Privitera, F.; Castello, G.; Lo Rizzo, C.; Mencarelli, M.A.; Pinto, A.M.; Ariani, F.; Currò, A.; Lamacchia, V.; Canitano, R.; et al. IQSEC2 disorder: A new disease entity or a Rett spectrum continuum? Clin. Genet. 2021, 99, 462–474. [Google Scholar] [CrossRef]
- Shoubridge, C.; Tarpey, P.S.; Abidi, F.; Ramsden, S.L.; Rujirabanjerd, S.; Murphy, J.A.; Boyle, J.; Shaw, M.; Gardner, A.; Proos, A.; et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat. Genet. 2010, 42, 486–488. [Google Scholar] [CrossRef] [Green Version]
- Gandomi, S.K.; Farwell Gonzalez, K.D.; Parra, M.; Shahmirzadi, L.; Mancuso, J.; Pichurin, P.; Temme, R.; Dugan, S.; Zeng, W.; Tang, S. Diagnostic exome sequencing identifies two novel IQSEC2 mutations associated with X-linked in-tellectual disability with seizures: Implications for genetic counseling and clinical diagnosis. J. Genet. Couns. 2014, 23, 289–298. [Google Scholar] [CrossRef]
- Helm, B.M.; Powis, Z.; Prada, C.E.; Casasbuenas-Alarcon, O.L.; Balmakund, T.; Schaefer, G.B.; Kahler, S.G.; Kaylor, J.; Winter, S.; Zarate, Y.A.; et al. The role of IQSEC2 in syndromic intellectual disability: Nar-rowing the diagnostic odyssey. Am. J. Med. Genet. A 2017, 173, 2814–2820. [Google Scholar] [CrossRef]
- Tzschach, A.; Grasshoff, U.; Beck-Woedl, S.; Dufke, C.; Bauer, C.; Kehrer, M.; Evers, C.; Moog, U.; Oehl-Jaschkowitz, B.; Di Donato, N.; et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 2015, 23, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Yohe, M.E.; Randazzo, P.A.; Gruschus, J.M. Allosteric properties of PH domains in Arf regulatory proteins. Cell. Logist. 2016, 6, e1181700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.M.; Price, E.M.; Jones, M.J.; Balaton, B.P.; Kobor, M.S.; Brown, C.J. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 2015, 24, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Moey, C.; Hinze, S.J.; Brueton, L.; Morton, J.; McMullan, D.J.; Kamien, B.; Barnett, C.P.; Brunetti-Pierri, N.; Nicholl, J.; Gecz, J.; et al. Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet. 2016, 24, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Gui, H.S.; Kwan, J.S.; Bao, S.Y.; Sham, P.C. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012, 40, e53. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | ND0673 | ND1443 |
|---|---|---|
| Variant (transcript) | NM_001111125: c.2459+21C>T | NM_001111125:3300dup |
| Variant (protein) | p.Pro745Valfs*12 | p.Leu1027Profs*79 |
| Mutation effect | Splicing | Frameshift |
| Inheritance | De novo | De novo |
| Patient’s gender | Male | Female |
| Age | 14 | 15 |
| Family history | Brother and parents healthy | Maternal cousins with intellectual disability and Down syndrome |
| Clinical features | ||
| Neurological manifestations | Intellectual disability | Intellectual disability |
| Seizures | Focal-onset seizure | |
| Global developmental delay | ||
| Epileptic encephalopathy | ||
| Abnormal pyramidal signs | ||
| Cognitive impairment | ||
| Ataxia | ||
| Spasticity | ||
| Motor delay | ||
| Generalized hypotonia | ||
| Unsteady galt | ||
| Difficulty walking | ||
| Speech deficits | Delay speech and language development | |
| Behavioral disturbances | Autistic behaviour | Autistic behavior |
| Obsessive-compulsive and aggressive behavior | ||
| Stereotypy | ||
| Hyperactivity | ||
| Low frustration tolerance | ||
| Impulsivity | ||
| Metabolic test | Normal | |
| Brain magnetic resonance | Normal in the first year | Normal |
| Discrete myelination delay at 2 y/o | ||
| Computed axial tomography | Normal | |
| Electroencephalogram | Abundant epileptic anomalies especially in the bilateral frontal region at 5 y/o | Normal at 9 years of age |
| Epileptic anomalies in the right anterior fronto-temporal region at 13 years of age | ||
| Other genetic studies | ||
| Kariotype | - | Normal |
| Array-CGH | Normal | Normal |
| Gene panel for epilepsy | Negative | |
| Mitochondrial DNA study | Normal | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baladron, B.; Mielu, L.M.; López-Martín, E.; Barrero, M.J.; Lopez, L.; Alvarado, J.I.; Monzón, S.; Varona, S.; Cuesta, I.; Cazorla, R.; et al. Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder. Int. J. Mol. Sci. 2022, 23, 9480. https://doi.org/10.3390/ijms23169480
Baladron B, Mielu LM, López-Martín E, Barrero MJ, Lopez L, Alvarado JI, Monzón S, Varona S, Cuesta I, Cazorla R, et al. Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder. International Journal of Molecular Sciences. 2022; 23(16):9480. https://doi.org/10.3390/ijms23169480
Chicago/Turabian StyleBaladron, Beatriz, Lidia M. Mielu, Estrella López-Martín, Maria J. Barrero, Lidia Lopez, Jose I. Alvarado, Sara Monzón, Sarai Varona, Isabel Cuesta, Rosario Cazorla, and et al. 2022. "Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder" International Journal of Molecular Sciences 23, no. 16: 9480. https://doi.org/10.3390/ijms23169480
APA StyleBaladron, B., Mielu, L. M., López-Martín, E., Barrero, M. J., Lopez, L., Alvarado, J. I., Monzón, S., Varona, S., Cuesta, I., Cazorla, R., Lara, J., Iglesias, G., Román, E., Ros, P., Gomez-Mariano, G., Cubillo, I., Miguel, E. H. -S., Rivera, D., Alonso, J., ... Martínez-Delgado, B. (2022). Differences in Expression of IQSEC2 Transcript Isoforms in Male and Female Cases with Loss of Function Variants and Neurodevelopmental Disorder. International Journal of Molecular Sciences, 23(16), 9480. https://doi.org/10.3390/ijms23169480